
Clinical Genetic Testing
In recent years, the value of genetic testing has increased for the diagnosis of various genetic disorders as seen by care-givers in OB/GYN, pediatrics, neurology and hematology/oncology. Whether used to identify chromosome abnormalities, characterize hematologic malignancies or determine the risk factors of inherited disease, clinical cytogenetics and DNA diagnostic laboratories have become key players in providing state-of-the art patient care.
The role of cytogenetics has grown since the discovery of trisomy 21 in Down syndrome and monosomy X in Turner syndrome nearly 40 years ago. Chromosome abnormalities have been associated with birth defects, mental retardation, infertility, pregnancy loss, hematologic disease and cancer. Prenatal cytogenetic analysis has become the standard of care for those patients at increased risk because of advanced maternal age, abnormal maternal serum screen, abnormal ultrasound findings, or a family history of birth defects. Many characteristic chromosome abnormalities have been associated with specific hematologic diseases and their identification provides prognostic and diagnostic information. Molecular cytogenetic techniques, especially fluorescent in situ hybridzation (FISH), have become an important adjunct to classical cytogenetics. Several chromosomal syndromes (e.g. Williams, Miller-Dieker and DiGeorge/ Velocardiofacial syndromes) have been identified that are at or beyond the limits of standard chromosome methods, and diagnosis is incomplete without both karyotype analysis and specific FISH assays. Using DNA probes, FISH allows for more sensitive and specific identification of gene sequences in interphase and metaphase cells.
The availability of diagnostic DNA analysis for inherited and acquired disorders has expanded dramatically. With the understanding of the genetic mechanisms of several diseases and syndromes, the DNA diagnosis of cystic fibrosis, sickle cell disease and other diseases can now be made. Carrier testing, presymptomatic DNA studies and prenatal diagnosis are available for many genetic diseases. Previously identified by cytogenetic analysis, patients with Fragile X, Prader-Willi and Angelman syndromes are now more accurately identified through DNA testing. The ability to detect alterations in oncogenes and tumor-suppressor genes is an invaluable tool in the diagnosis and treatment of cancer patients. Based on a patient’s family history, a patient’s predisposition to certain genetic disorders can be determined.
The combination of an informed physician, genetic testing, counseling, and treatment or supportive care provides a patient and their family with the most effective means of coping with a genetic disorder or birth defect.
FISH
Fluorescent in situ hybridization (FISH) has proven to be a valuable tool in the identification of chromosome abnormalities and is regarded as an important adjunct to karyotype analysis. FISH techniques allow for the highly sensitive detection of specific nucleic acid sequences in both metaphase and interphase cells. FISH has become the method of choice in identifying several of the microdeletion syndromes, including DiGeorge/Velocardiofacial (VCFS), Williams, Miller-Dieker, and Smith-Magenis. Testing of uncultured amniocytes for aneuploidy of chromosomes 13, 18, 21, X and Y provides a rapid screening tool for at-risk pregnancies. The technique is also used to determine the origin of marker chromosomes or to characterize complex rearrangements. Probes are available to detect leukemia associated aneuploidy (e.g. +12 in CLL), chromosome rearrangements, and to identify oncogene amplification (e.g. n-myc in neuroblastomas).
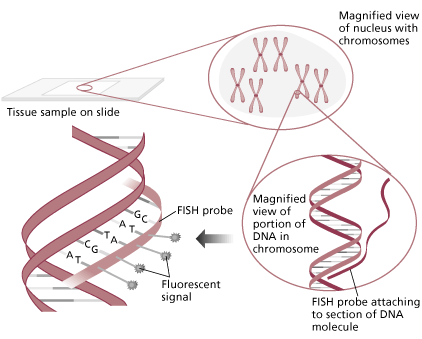
Graphic provided by Vysis, Inc.
Several types of DNA probes have been developed and each has its unique applications. Repetitive sequence probes (usually centromere specific), are useful in determining ploidy and identifying small marker chromosomes. Chromosome paints consisting of a cocktail of probes specific for a particular chromosome are used to identify translocation segments. Single gene probes contain specific regions of DNA that contain the critical region known to be duplicated, deleted or translocated in a particular syndrome or leukemia. Examples of these probes include the DiGeorge syndrome probe used to identify a deletion of 22q11.2 or the PML/RARA probe associated with t(15;17). Of recent interest is the identification of submicroscopic telomere deletions or rearrangements and their association with mental retardation.
When it’s protocol:
FISH testing is performed on request for the detection of specific microdeletion syndromes. It is the protocol of the laboratory that if the reason indicated for cytogenetic analysis (or the patient’s clinical description) are pertinent, FISH analysis is performed in addition to the routine G-banded karyotype. For example, in all cases of chronic lymphocytic leukemia, FISH is used to identify a trisomy 12 cell population. Likewise, when a structural heart defect is identified prenatally by ultrasound, we employ FISH to identify the common 22q deletion associated with VCFS.
Fragile X Syndrome: Clinical and Molecular Features
Fragile X syndrome is the most common inherited form of mental retardation. It is characterized by mental retardation of variable severity and distinctive physical and behavioral characteristics that affect both males and females (Table 1). In males, dysmorphic features generally appear in childhood and include: macrocephaly; a long thin face; large, prominent ears, forehead and jaw; high arched palate and lax muscle tone. Some patients may have hyperextensible joints, flat feet and mitral valve prolapse. Macro-orchidism begins to develop in early puberty and is present in almost all postpubertal males.
| Table 1 Clinical Features of Fragile X Syndrome in Males | |
| Physical Features | Behavioral Characteristics |
| Prominent, large ears | Development delay |
| Long, narrow face | Mental retardation |
| Prominent forehead | Learning disabilities |
| Prominent, square chin | Hyperactivity |
| High-arched palate< | Attention deficit disorder |
| Macrocephaly | Speech perseveration |
| Hand calluses | Autistic-like features including hand biting, hand flapping, and poor eye contact |
| Mitral valve prolapse | Shyness, social anxiety |
| Macro-orchidism | Tactile defensiveness |
| Seizures | Frequent temper tantrums, mood instability |
| Hyperextensible finger joints | Difficulty adjusting to change |
| Flat feet |
Mental retardation is common but varies in severity in affected males. Greater than 90% of affected males are mentally retarded; the average IQ of males with a completely methylated full mutation is between 40-45. Approximately 20% of males have seizures. Behavioral manifestations in affected males include hyperactivity, attention problems, language delays, poor eye contact, temper tantrums and perseverative speech. The majority of boys display autistic-like behavior, such as hand flapping, hand biting and tactile defensiveness.
Females with fragile X syndrome are usually less severely affected than males because they have two X chromosomes, only one of which carries the fragile X mutation. Females with a full fragile X mutation demonstrate an extremely variable phenotype. Approximately 50-70% of females with the full mutation have an IQ of 84 or less. Girls with a full mutation and fragile X syndrome usually present with learning disabilities, including math difficulties, language deficits and attention deficit disorders. Some of the characteristic facial features, such as prominent ears, may be present in females.
Fragile X syndrome results from a complex interaction between alterations in the length of a trinucleotide repeat, (CGG)n, and changes in the expression of the FMR-1 (fragile X mental retardation) gene. The CGG repeats are located within the 5′ untranslated region of the FMR-1 gene. In normal individuals, the number of repeats ranges between ~6 and ~50. These alleles are inherited stably in a Mendelian manner. There appears to be a threshold of ~50 repeats, above which the DNA segment becomes unstable and is subject to DNA expansion. FMR-1 alleles with between ~50 to ~200 are termed premutations. FMR-1 premutation genes have normal transcription and translation and no phenotypic effects appear in premutation carriers. Thus a female or male may carry a premutation and be clinically unaffected.
In fragile X syndrome, the number of repeats is expanded to greater than 200 units. This is called a full mutation. Males with a full mutation have the characteristic phenotype while half of females with a full mutation are mentally impaired. The cytosine bases in the CG dinucleotides in the triplet repeat and the associated CpG island found in the 5′ untranslated region of the FMR-1 gene becomes hypermethylated when the number of repeats exceeds 200. This methylation results in the transcriptional and translational suppression of FMR-1 (Figure 1). Therefore, most affected males produce no FMR-1 protein. It is the lack of protein which is the critical factor in the pathogenesis of the fragile X phenotype. This is best seen in males who are mosaic for a premutation and full mutation who demonstrate partial FMR-1 methylation and milder clinical involvement.
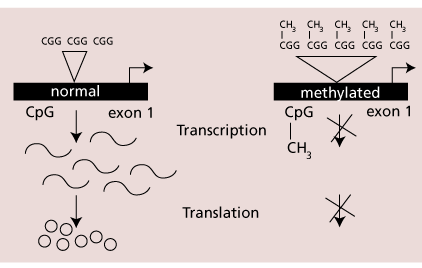 |
The CGG repeats in a female FMR-1 premutation gene can undergo expansion and be passed onto an offspring who may inherit a full mutation and be affected with fragile X syndrome. The length of the expansion to a full mutation is low for small premutations but increases to nearly 100% as the number of repeats approaches 90. A male carrying a premutation ranging in size from 50-200 repeats is a normal transmitting male. The premutation is stable during male gametogenesis, therefore, males transmit a premutation to all of their daughters, but to none of their sons (since males pass their X chromosome to daughters and their Y chromosome to sons).
| Table 2 Fragile X DNA Testing Should be Considered in the Following Individuals |
| Males or females with mental retardation, developmental delay, or autism, especially if they have physical or behavioral characteristics of fragile X syndrome, have a family history of fragile X syndrome, or have relatives with undiagnosed mental retardation. |
| Individuals seeking reproductive counseling who have a family history of fragile X syndrome or a family history of undiagnosed mental retardation. |
| Patients with a cytogenetic fragile X test result discordant with their phenotype, especially if they have a strong clinical indication, have an ambiguous cytogenetic test result, or have an atypical phenotype with a positive cytogenetic test result. |
| Fetuses of known carrier mothers. |
| Population screening is not recommended, unless it is part of a well defined clinical research protocol. |
DNA testing replaced cytogenetic fragile X testing as the diagnostic method of choice, except in rare circumstances. Recommendations for testing have been published by the American College of Medical Genetics (Table 2). The strategy used for molecular diagnosis of fragile X syndrome in our laboratory uses the polymerase chain reaction (PCR) and Southern blot-based direct detection of FMR-1 allele sizes and methylation status. PCR is used to obtain an accurate determination of the CGG repeat number in normal and premutation carrier individuals. Determination of the exact repeat number is important because it can drastically alter the estimate of a female’s risk of producing affected offspring and is important in genetic counseling. However, repeats of greater than ~125 units are generally difficult to amplify by PCR and will usually give a null PCR product. Southern blot analysis is useful for estimating the number of CGG repeats in premutation and full mutation alleles.
Microdeletion Syndromes
Since first being described in 1986, the number of microdeletion/microduplication syndromes has increased and the methodology for identification has evolved. The term “contiguous gene syndrome” defines a group of disorders with microdeletions or microduplications of chromosome segments associated with gain or loss of single genes. While several of these syndromes were initially identified through cytogenetic analysis, not all have a visible cytogenetic abnormality and molecular techniques can now be used to facilitate their detection.
Clinical features for the more commonly seen microdeletion syndromes are listed in the table. In addition to high-resolution karyotype analysis, fluorescent in situ hybridization techniques are routinely used to identify patients with these syndromes. The diagnosis of Prader-Willi and Angelman syndromes involves molecular testing for parent-of-origin dependent DNA methylation sites.
Features of Microdeletion syndromes
| FISH and Karyotype Analysis | |
| Wolf Syndrome (4p-) | pre and post natal growth retardation severe mental retardation severe heart malformations microcephaly cleft lip and palate broad nasal root |
| Cri du chat (5p-) | cat cry in infancy mental retardation low birth weight/failure to thrive microcephaly round face hypertelorism |
| Williams syndrome (7q11.23) | elfin-like facies dental anomalies mental retardation growth deficiency congenital heart defect infantile hypercalcemia |
| Smith-Magenis syndrome (17p11.2) | moderate mental retardation prominent forehead flat, broad midface self-mutilating behavior disturbed REM sleep |
| Miller-Dieker syndrome (17p13) | microcephaly Type I lissencephaly heart & kidney defects seizures/abnormal EEG prominent forehead vertical skin furrowing brow |
| DiGeorge syndrome (22q11.2) | thymic hypolplasia cellular immune deficiency parathyroid hypoplasia hypocalcemia conotruncal heart defects low set ears hypertelorism |
| Velocardiofacial syndrome (22q11.2) | cleft palate congenital heart disease any conotruncal heart defect mild mental retardation prominent nose hyperextensible hands & fingers |
| CHARGE Sequence (22q11.2) | colomba heart defects atresia choanae mental retardation genital hypoplasia ear anomalies & deafness postnatal growth deficiencies |
| DNA Testing | |
| Angelman syndrome (15q11.2) | developmental delay seizures microcephaly inappropriate laughter stiff, ataxic gait |
| Prader-Willi syndrome (15q11.2) | mental retardation hypotonia & failure to thrive in infancy obesity & hyperphagia in childhood hypogonadism |
Importance of Request Form Information
Genetic analysis is a highly specialized form of testing and a complete description of a patient’s clinical features or suspected diagnosis is critical in providing an accurate and informative report.
While routine karyotype testing involves analysis of several cells looking for abnormalities in chromosome number or structure pattern, accurate clinical information can pinpoint particular chromosomes of interest. A clearly defined reason for karyotyping, including a description of the patient’s physical characteristics, helps the laboratory focus on certain regions of chromosomes known to be associated with a defined syndrome or leukemia and may also indicate that additional testing is required. For example:
- Features including cleft palate, Ventricular Septal Defect (VSD), and mental retardation (MR) are associated with DiGeorge/Velocardiofacial syndrome (deletion of 22q11.2) and warrant FISH testing in addition to routine karyotype analysis.
- In leukemia studies, additional cell cultures are initiated if a B-cell disorder is suspected. An indication of “r/o leukemia” rather than “r/o lymphoma” or “r/o CLL” may preclude identification of an abnormal clone, because lymphoma studies include evaluation of cells from B-cell stimulated culture, and CLL studies include FISH for a slowly proliferating +12 population.
- An indication of “family history of chromosome abnormality” is best investigated if the abnormality is indicated or previous cytogenetic reports are included with the request. Some rearrangements are very subtle, so providing specific information is essential.
- In cases where mosaicism is a common feature, additional cells are analyzed. Typical analysis involves examination of 20 metaphase cells, whereas 30 cells are examined if mosaicism is suspected (e.g. sex chromosome abnormalities, presence of contractures, infertility).
In the DNA Diagnostic Laboratory, a pertinent family history or pedigree and medical history is essential in our providing an accurate interpretation in the patient’s final report. Based on the history reported, different recommendations may be made regarding additional family members who may benefit from DNA studies or genetic counseling or additional studies that may directly benefit the patient. Information regarding the patient’s age or date of birth, race or ethnic background, and first date of last menstrual period (if patient is pregnant) will ensure that the most accurate interpretation of the results is provided. For example:
- Symptomatic patient versus asymptomatic patient with a positive family history: A patient with a history of venous thrombosis and a negative DNA study for Factor V (Leiden) may benefit from a recommendation for additional DNA studies (Prothrombin and Methylenetetrahydrofolate reductase) while such testing would not be indicated in an asymptomatic patient who is being tested for Factor V (Leiden) because of a first-degree relative with the mutation.
- Rule-out carrier versus rule-out affected: A pregnant woman of normal intellect who has a brother with mental retardation and a pregnant woman with a learning disability require different interpretations of their results, even if they both have normal fragile X syndrome DNA studies.
- Importance of race/ethnic background: A negative hereditary hemochromatosis DNA study has different implications for a Caucasian patient as than for an African American patient because there are differences in the normal and mutant allele frequencies in these two populations. Different interpretations are provided for these ethnic backgrounds.
 |
Testing Services
The Cytogenetics Laboratory at Henry Ford Hospital is a division of the Department of Medical Genetics and is a major reference laboratory for physicians and hospitals in the Midwest. We welcome telephone or email consultations.
- Karyotype Analysis and FISH analysis available on:
amniotic fluid
chorionic villi
peripheral blood
products of conception
skin biopsy
bone marrow aspirate
lymph node
pleural effusion
solid tumor - Verbal or faxed preliminary reports on:
90% of amniotic fluid
specimens within 5 days
abnormal infant peripheral
blood specimens within
48 hours
leukemic specimens within
24-48 hours
The DNA Diagnostic Laboratory at Henry Ford Hospital is a division of the Department of Medical Genetics. The laboratory provides service to physicians and hospitals inside and outside of the Henry Ford Health System. The laboratory routinely employs current molecular genetic techniques for the diagnosis of specific genetic disorders listed below. Testing for additional genetic diseases will be offered as appropriate. If there is a specific genetic disease or test which is not currently offered, but one that you are interested in, please let us know. Consultation with the Laboratory Director is always welcome.
Karyotype Analysis and FISH analysis available on:
- Cystic Fibrosis
- Cystic Fibrosis PolyT Allele
- Factor V (Leiden)
- Factor II (Prothrombin)
- Methylenetetrahydrofolate reductase (MTHFR)
- Coagulation Panel (Includes Factor V, Prothrombin, MTHFR)
- Hereditary Hemochromatosis
- Hereditary Pancreatitis
- Sickle Cell Anemia / Hemoglobin C Disease
- Prader-Willi Syndrome
- Angelman Syndrome
- Fragile X Syndrome
- Myotonic Dystrophy
- Multiple Endocrine Neoplasia Type 2A (MEN2A)
- Multiple Endocrine Neoplasia Type 2B (MEN2B)
- Familial Medullary Thyroid Carcinoma (FMTC)
- DNA Extraction
| Daniel L. Van Dyke, Ph.D. Cytogenetics Laboratory | dvandyk1@hfhs.org | |
| Kristin G. Monaghan, Ph.D. DNA Diagnostic Laboratory | kmonagh1@hfhs.org | |
| Jacquelyn R. Roberson, M.D. Genetics Clinic | jrobers1@hfhs.org | |
| Anne Wiktor FISH Laboratory | awiktor1@hfhs.org | |
| Juanita Clark Laboratory reports; billing | jclark1@hfhs. | |