
Clinical Evaluation of Hemoglobinopathies: Part I. Thalassemia
Hemoglobin is a remarkable molecule that provides erythrocytes with the ability to carry oxygen to tissues and to remove carbon dioxide that is generated as a metabolic product. Because of this critical function, significant alterations in either the structure or the amount of the hemoglobin molecule produced may have profound effects on the patient. This article reviews the most common alterations encountered clinically and the laboratory testing for streamlining the clinical diagnosis.
Background on Hemoglobin Structure
Hemoglobin molecules are composed of two pairs of globin chains each containing a heme group at its core. The heme group is where the exchange of oxygen and carbon dioxide occurs. Although mutations may occur at many sites in the hemoglobin molecule, the biological function of hemoglobin is usually most severely affected by alterations that distort the three-dimensional structure in this core location.
Hemoglobin Production
The genetic information for hemoglobin production resides on two chromosomes. The alpha group of genes exists as duplicated sets on chromosome 16 and the beta group of genes (including delta and duplicated gamma) are located on chromosome 11. Two types of genetic information are present. One determines the amino acid sequence of the hemoglobin molecules and the other regulates how much hemoglobin is produced.
Figure1A. Production of globin chains. Sequence of globin chain synthesis in the embryo and fetus. Note that the embryonic globin chains cease production by the end of the first trimester. The ratio of ![]() and
and ![]() is listed above the total of gamma chains.
is listed above the total of gamma chains.
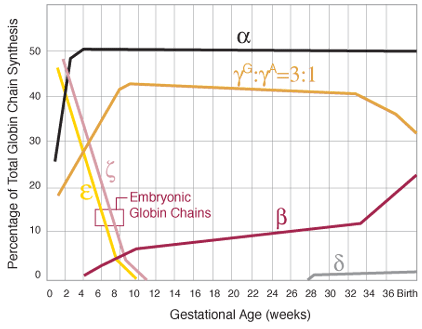
Early-on during embryonic life, zeta and epsilon chains are synthesized resulting in the embryonic hemoglobins Gower 1 ![]() , Gower 2
, Gower 2 ![]() , and Portland
, and Portland ![]() . After the first two months of gestation, a switch occurs on chromosome 16 that turns off the production of zeta chains and begins production of alpha chains. Similarly, at this time, the synthesis of epsilon chains by chromosome 11 stops and production of gamma chains begins resulting in formation of fetal hemoglobin (HbF)
. After the first two months of gestation, a switch occurs on chromosome 16 that turns off the production of zeta chains and begins production of alpha chains. Similarly, at this time, the synthesis of epsilon chains by chromosome 11 stops and production of gamma chains begins resulting in formation of fetal hemoglobin (HbF) ![]() . Because of this process, by the tenth week of gestation, HbF is the predominant oxygen-carrying molecule (figure 1A). HbF has a higher affinity for oxygen than adult hemoglobin, a characteristic that facilitates transport of oxygen across the placenta [1].
. Because of this process, by the tenth week of gestation, HbF is the predominant oxygen-carrying molecule (figure 1A). HbF has a higher affinity for oxygen than adult hemoglobin, a characteristic that facilitates transport of oxygen across the placenta [1].
Figure1B. Production of globin chains. Sequence of globin chain synthesis during the first year of life. Note the change in the ratio of ![]() and
and ![]() .
.
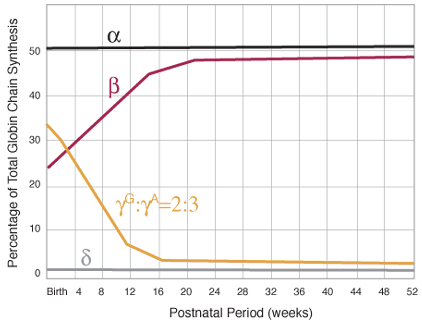
During the fifth month of fetal life, a gradual increase in production of beta chains occurs. Shortly before birth, a switch occurs that shuts off the production of gamma chains by most erythroid cells and begins synthesis of beta and small amounts of delta chains from the genetic information on chromosome 11 (figure 2). This results in the production of adult hemoglobins: A ![]() and A2
and A2 ![]() . During adult life, only a small amount of HbF is made under normal circumstances. In the fetus, HbF is produced in all erythrocytes, whereas its production in adults is restricted to “F-cells”. Structurally F-cells resemble adult rather than fetal cells, but they produce both adult hemoglobin and HbF. In times of stress to the erythroid compartment such as anemia, leukemia, response to iron therapy in an iron-deficient patient, the number of F-cells increases and the amount of HbF increases. Our normal adult ranges for hemoglobins are shown in Table 1.
. During adult life, only a small amount of HbF is made under normal circumstances. In the fetus, HbF is produced in all erythrocytes, whereas its production in adults is restricted to “F-cells”. Structurally F-cells resemble adult rather than fetal cells, but they produce both adult hemoglobin and HbF. In times of stress to the erythroid compartment such as anemia, leukemia, response to iron therapy in an iron-deficient patient, the number of F-cells increases and the amount of HbF increases. Our normal adult ranges for hemoglobins are shown in Table 1.
Table 1. Normal Ranges of Adult Hemoglobins
| Hemoglobin | Globin Chain Composition | Adult Concentration* |
| HbA | 96-98% | |
| HbA2 | 2.3-3.5% | |
| HbF | < 2% |
| *Adult concentrations are usually attained by 6-8 months of age. |
Hemoglobinopathies
Clinical Information.
When a patient is evaluated for the presence of a hemoglobinopathy, the laboratory needs to know several pieces of clinical information to optimize the interpretation (Table 2). First we need to know the date of birth because HbF values differ considerably in the first few months of life. In addition to HbF, until adolescence, there is a continuous change in the CBC parameters that are used to determine if a decrease in such parameters as HbA2 represents iron deficiency versus alpha thalassemia. If CBC information is not provided, the evaluation by our preferred screen, high-pressure liquid chromatography (HPLC), and/or gel-electrophoresis is incomplete. As will be made clear below, without this information, some conditions such as heterozygous thalassemia and high-oxygen affinity hemoglobins cannot be confirmed, and in some cases will not be suspected.
Another important piece of information is if the patient has received a recent blood transfusion, and if so how much. A blood transfusion will alter the percentage of any variant hemoglobin that is present and may also mask the presence of alpha thalassemia (by increasing the HbA2) or beta thalassemia (by decreasing the percent of HbA2).
Lastly, when possible, the country of origin of the patient’s ancestors can play an important role in the evaluation of a difficult case. There are literally hundreds of known genetic alterations that may occur. Knowing the country of origin of the patient or the patient’s ancestors may streamline an otherwise expensive evaluation.
Table 2. Clinical Information Needed for Optimal Interpretation
| Clinical Information Needed | How the Information is Used |
| Birth date of Patient | 1. Hemoglobin F varies during the first year of life. 2. CBC parameters vary considerably until adolescence. |
| Sex of patient | CBC parameters vary by sex |
| CBC | Information from the CBC is important to detect/confirm the presence of some forms of thalassemia and to help distinguish thalassemia from iron deficiency |
| Transfusion History | This information is critically important to distinguish between heterozygous and homozygous variants. For instance, the hemoglobin electrophoresis (or HPLC) on blood from a transfused patient with homozygous sickle cell disease may resemble sickle cell trait. |
| Ancestral Country of Origin | Knowing the country where the patient or the patient’s ancestors originated may be helpful in the differential diagnosis of unusual hemoglobinopathies or some of the forms of thalassemia. |
There are two large categories of abnormalities that may compromise hemoglobin function: thalassemia (decreased production of usually normal hemoglobin molecules) and structural abnormalities.
Thalassemia
A Detroit native, Dr. Thomas Cooley and his colleague Dr. Pearl Lee reported cases of severe anemia in children in the Mediterranean region in 1925 [1]. Because the cases were located near the Mediterranean Sea, Cooley and Lee coined the term “Thalassemia” from the Greek “thalassa” for “sea”. These severely affected patients represent Thalassemia Major. Later, milder forms of this anemia became apparent that were termed Thalassemia Intermedia and carriers (Thalassemia Minor) of the condition are clinically silent with minor, or in some cases, no hematological abnormalities. Three major categories of Thalassemia exist.
Alpha Thalassemia
Alpha Thalassemia is due to the decrease in production of structurally normal alpha globin chains. As mentioned above, alpha globin is synthesized by four alpha globin genes on Chromosome 16 (two alpha globin genes on each chromosome). The main cause of decreased production is deletion of one or more alpha genes. Molecular studies have characterized the most common forms of deletions (Table 3) [2]. Less commonly, point mutations in regulatory genes will result in a decrease or absence of production by one or more alpha genes. About 30 such mutations and small deletions/insertions have been reported. Point mutations are more common, however, in Beta Thalassemia .
Table 3. Common Forms of Deletional Alpha Thalassemia
| Thalassemia Deletion | Ancestral Geographic Origin | Clinical Features |
| Africa, Asia, Mediterranean, Pacific Islanders | Heterozygotes-normal erythrocyte parameters | |
| Mainly Asia | Heterozygotes-mild microcytosis | |
| Southeast Asia deletion | Asia, Mediterranean | Heterozygotes- microcytosis |
Alpha Thalassemia Minor
Loss of one alpha gene (termed Alpha Thalassemia-2) may result in an MCV in the low normal to slightly low range. The amount of reduction in alpha globin produced will depend upon the specific deletion. For instance, with the ![]() -3.7 deletion, there is a compensatory increase in the production of alpha globin by the remaining alpha gene. This produces a silent carrier. With the
-3.7 deletion, there is a compensatory increase in the production of alpha globin by the remaining alpha gene. This produces a silent carrier. With the ![]() -4.2 deletion, there is a greater loss of alpha gene production, however, clinically, the patient is unaffected. If two alpha genes are deleted (Alpha Thalassemia-1), the MCV will almost always be reduced. There also may be a minor reduction in the MCHC, the erythrocyte count may be in the high normal range or slightly increased, while the RDW remains in the normal range (unless there is a co-existing iron deficiency). A neonate with Alpha Thalassemia-1 may have small amounts of Hemoglobin Barts
-4.2 deletion, there is a greater loss of alpha gene production, however, clinically, the patient is unaffected. If two alpha genes are deleted (Alpha Thalassemia-1), the MCV will almost always be reduced. There also may be a minor reduction in the MCHC, the erythrocyte count may be in the high normal range or slightly increased, while the RDW remains in the normal range (unless there is a co-existing iron deficiency). A neonate with Alpha Thalassemia-1 may have small amounts of Hemoglobin Barts ![]() , but the amount is smaller than in neonates who will develop Hemoglobin H disease. In addition, examination of the hemoglobin by HPLC demonstrates HbA2 that is either decreased or in the lower end of the normal range [2].
, but the amount is smaller than in neonates who will develop Hemoglobin H disease. In addition, examination of the hemoglobin by HPLC demonstrates HbA2 that is either decreased or in the lower end of the normal range [2].
Hemoglobin H Disease
The loss of three of the four alpha genes results in a marked decrease in the production of alpha globin. However, the production of beta chains (in the adult) and gamma chains (in the fetus) are unaffected. As a result, excessive beta chains and gamma chains are synthesized in the adult and fetus respectively. In the fetus, the excess gamma chains combine to form Hb Barts ![]() and in the adult, the excess beta chains combine to form Hb H
and in the adult, the excess beta chains combine to form Hb H ![]() which are both ineffective in carrying oxygen and which damage the erythrocytes in which they reside. A loss of three alpha genes results in Hemoglobin H disease. This disease varies considerably from one patient to another in terms of its clinical severity. Some patients are clinically well, although there are always notable hematological and HPLC changes: low MCV, low MCHC, low hemoglobin concentration, elevated erythrocyte count, decreased HbA2. Other cases have a transfusion-dependent anemia. In the latter case, chelation therapy is needed to prevent iron overload [3].
which are both ineffective in carrying oxygen and which damage the erythrocytes in which they reside. A loss of three alpha genes results in Hemoglobin H disease. This disease varies considerably from one patient to another in terms of its clinical severity. Some patients are clinically well, although there are always notable hematological and HPLC changes: low MCV, low MCHC, low hemoglobin concentration, elevated erythrocyte count, decreased HbA2. Other cases have a transfusion-dependent anemia. In the latter case, chelation therapy is needed to prevent iron overload [3].
The variability seen in Hemoglobin H disease results from the different genetic causes for the decreased production of alpha globin chains and from the presence of other abnormalities which may ameliorate the effects of the excess beta or gamma globin chains. For instance, if an individual with loss of three alpha genes also has decreased production of beta genes due to the co-existence of beta thalassemia, there will not be as many free beta globin chains to form HbH that can damage cells. This will result in a milder form of Hemoglobin H disease.
Because alpha globin is a component of HbF, the newborn fetus with a three alpha gene deletion will have detectable manifestations: low MCV, low MCHC, low hemoglobin, increased erythrocyte count. Although HbA2 will be decreased, too little HbA2 is present at birth to make a decrease in this hemoglobin obvious. However, HPLC or gel elctrophoresis will demonstrate the presence of Hb Barts and occasionally Hb H. The amount of Hb Barts present at birth may be useful in predicting the severity of the HbH disease that the child will develop.
If all four alpha genes are deleted, the poor oxygen-carrying capacity of hemoglobin Barts is insufficient for survival of the fetus.
Beta Thalassemia
Beta Thalassemia is due to the decreased production of beta globin. The beta globin genes occur singly on chromosome 11 (figure 2). In contrast to Alpha Thalassemia where large deletions cause the problem, the most common cause of decreased production of beta globin is single gene mutation rather than deletion of chromosomal material. Although the current diagnosis of Beta Thalassemia depends on a combination of hemoglobin electrophoresis and clinical information, molecular techniques are being developed that will eventually lead to identification of the exact mutation resulting in a more precise prediction of the consequences to the patient and the patient’s offspring. Because over 200 mutations of the beta genes or beta gene production controlling regions have been described, there is great heterogeneity in the clinical and laboratory picture of patients with Beta Thalassemia. Less commonly, chromosomal deletions rather than mutations can cause Beta Thalassemia [1, 4].
Figure 2. Beta Globin Gene Cluster
(Short Arm of Chromosome 11) Beta globin genes on chromosome 11. The order of the structural genes reflects their translation during embryonic, fetal and postnatal life with the exception of ![]() which is a vestigial gene.
which is a vestigial gene.
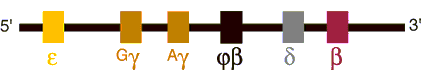
Beta Thalassemia Minor and Intermedia
When the mutation causes a decrease in production of beta globin by one beta gene, it is termed Beta+ Thalassemia. The amount of decrease in production varies with the type of mutation. In general, Beta+ Thalassemias from individuals whose ancestors originated from the Mediterranean region are relatively severe producing marked decreases in the synthesis of beta globin, whereas, Beta+ Thalassemias from individuals whose ancestors originated in Africa tend to be mild. Mutations that completely eliminate production of beta globin by one gene are termed Betaº Thalassemia.
Clinically, patients with heterozygous Beta Thalassemia (whether Beta+ or Betaº ) are well because the production of beta globin by the unaffected beta gene is normal. Nonetheless, they have detectable hematological manifestations: low MCV, low MCHC, normal to slightly elevated erythrocyte counts, and often normal hemoglobin. By HPLC, we can accurately quantify the HbA2 that typically is increased in heterozygous Beta Thalassemia (figure 3). When the clinical information is combined with HPLC results, a diagnosis of heterozygous Beta Thalassemia Minor can be made. In addition to the increase in HbA2, some cases of heterozygous Beta Thalassemia Minor have an increase in the production of HbF.
| Figure 3. HPLC pattern of hemoglobin from a patient with heterozygous Beta Thalassemia. Note increased HbA2 (normal range 2.2-3.5). |
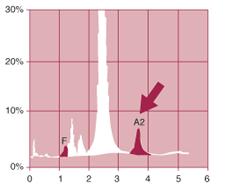 | Analyte ID%TimeAreaF1.11.221163P25.31.41106107P33.91.7177801Ao83.62.411661227A26.23.66104612Total Area1970910 1.1%A26.2% |
Because of the wide variety of genetic types of Beta Thalassemia, inheritance of two defective genes for Beta Thalassemia may result in a spectrum of clinical manifestations from mild anemia (for instance inheritance of two Beta+ genes) to Thalassemia Intermedia (inheritance of one Beta+ African and one Betaº gene, or two Beta+ Mediterranean genes) to severe transfusion dependent disease (inheritance of two Betaº genes). Patients with Beta Thalassemia Intermedia usually do not require transfusion, whereas those with Beta Thalassemia Major do.
Beta Thalassemia Major
The presence of Beta Thalassemia Major results in an absence of production of beta globin. Despite a modest compensatory increase in production of gamma globin genes, alpha globin is present in excess because its synthesis is unimpaired. The four alpha globin chains form an unstable molecule that precipitates in the erythrocytes during production damaging these cells and resulting in ineffective erythropoiesis [1, 4].
In an attempt to respond to the resulting anemia, there is marked expansion of the marrow (up to ten-fold) creating physical distortions in the skull and hands of patients with Thalassemia Major. Further, the marrow hyperplasia is accompanied by increased iron absorption with deposition of the iron in tissues. Early chelation therapy is critically important to prevent iron overload.
Because beta globin is a minor part of hemoglobin production in the fetus, CBC parameters may be normal at birth by virtue of the presence of HbF. But in Beta Thalassemia Major, the HPLC pattern in the newborn will disclose the complete lack of HbA that points to the underlying problem. HbF production markedly drops off after birth and since HbA cannot be produced in these children, Beta Thalassemia Major presents during the first year of life with severe anemia, low MCV, absence of HbA and elevated HbF. Confirmation of the diagnosis is made by performing family studies on parents, both of whom will have Beta Thalassemia Trait.
As with Hemoglobin H Disease, co-inheritance of other genetic problems may ameliorate some of the clinical symptoms. The presence of Alpha Thalassemia trait results in a decrease in production of the excess alpha chains thereby decreasing the damage that occurs during erythropoiesis.
Delta-Beta Thalassemia (“F” Thalassemia)
Delta-Beta Thalassemia results from a deletion in both the delta and beta genes on chromosome 11(figure 2). The gamma genes on the affected chromosome increase their production of gamma globin thereby increasing the amount of HbF. Delta-Beta Thalassemia heterozygotes clinically have Thalassemia Minor. The patients are clinically well, but may manifest a low normal or slightly low MCV and low MCHC on their hemogram. Usually one can detect a decrease in HbA2 (because the delta gene has been deleted from chromosome 11 and an increase in the amount of HbF. Because of increased production of HbF, Delta-Beta Thalassemia often is referred to as “F” Thalassemia [5].
Homozygous Delta-Beta Thalassemia may give a clinical picture of Thalassemia Intermedia with a mild anemia, low MCV, low MCHC, high normal to elevated erythrocyte count and a normal RDW (unless there is a concomitant iron deficiency). The HPLC pattern demonstrates HbF only. Because the total amount of gamma chains are inadequate to combine with the excess alpha chains, problems in erythrocyte production may occur, similar to, but not as severe as homozygous Betaº Thalassemia. Heterozygotes for Delta-Beta Thalassemia and Betaº Thalassemia can, however, present with as severe a disease as homozygous Betaº Thalassemia [6].
Hereditary Persistence of Fetal Hemoglobin (HPFH) may be confused with Delta-Beta Thalassemia because both result in an increased percentage of HbF in adult blood. The key difference between HPFH heterozygotes and Delta-Beta Thalassemia heterozygotes is that the amount of total hemoglobin in the latter is decreased resulting in a low MCV, low MCHC, high normal to elevated erythrocyte counts and often low or low normal HbA2. In contrast, in HPFH the total amount of hemoglobin produced is sufficient to prevent these abnormalities [6].
Production of HbF depends on the two gamma chain genes which are present on each chromosome 11 (figure 2). They differ by the production of alanine at position 136 on one (termed ![]() ) and glycine on the other (termed
) and glycine on the other (termed ![]() ) (Figure 1B). HPFH may result from a deletion that usually includes the delta and beta genes. However, HPHF may also result from point mutations on an otherwise intact chromosome 11 (non-deletional form). The amount of HbF produced varies from as little as 2% in one of the non-deletional forms of the condition to as much as 30% in a deletional heterozygote. Further, patients who are doubly heterozygous for HPFH and for variant genes such as for sickle hemoglobin will produce even higher percentages of HbF. This is due to the preferential binding of alpha globin chains to the normal gamma globin chain versus the abnormal sickle beta globin chain. Lastly, patients with homozygous HPHF produce only HbF and are clinically well with normal hemograms [6].
) (Figure 1B). HPFH may result from a deletion that usually includes the delta and beta genes. However, HPHF may also result from point mutations on an otherwise intact chromosome 11 (non-deletional form). The amount of HbF produced varies from as little as 2% in one of the non-deletional forms of the condition to as much as 30% in a deletional heterozygote. Further, patients who are doubly heterozygous for HPFH and for variant genes such as for sickle hemoglobin will produce even higher percentages of HbF. This is due to the preferential binding of alpha globin chains to the normal gamma globin chain versus the abnormal sickle beta globin chain. Lastly, patients with homozygous HPHF produce only HbF and are clinically well with normal hemograms [6].
Treatment of Thalassemia
Thalassemia minor and most cases of Thalassemia Intermedia do not require transfusion therapy. Yet, Thalassemia Major, whether due to homozygous Betaº Thalassemia or some (not all) HbH disease, requires careful treatment with a combination of transfusion and chelation therapy. The chelation therapy is extremely important because cases of Thalassemia Major may have clinically significant iron overload by the first year or two of life. In the past few years, hydroxyurea, sometimes together with recombinant erythropoietin, has been used to encourage the production of larger amounts of HbF which ameliorates the disease [7, 8]. By raising the level of HbF, some patients with Thalassemia Major and others with homozygous sickle disease have had transfusion requirements dramatically reduced. The use of bone marrow and stem cell transplants have been reported with some success [9, 10]. Unfortunately, such transplants are subject to severe problems with Graft-versus-Host disease and are not yet considered standard therapy.
References
- Olivieri, N.F., The beta-thalassemias. N Engl J Med, 1999. 341(2): p. 99-109.
- Steinberg, M.H., The interactions of alpha-thalassemia with hemoglobinopathies. Hematol Oncol Clin North Am, 1991. 5(3): p. 453-73.
- Kushner, J.P., J.P. Porter, and N.F. Olivieri, Secondary iron overload. Hematology (Am Soc Hematol Educ Program), 2001: p. 47-61.
- Schrier, S.L., Pathophysiology of thalassemia. Curr Opin Hematol, 2002. 9(2): p. 123-6.
- Galanello, R., et al., Homozygosity for nondeletion delta-beta(0) thalassemia resulting in a silent clinical phenotype. Blood, 2002. 100(5): p. 1913-4.
- Rochette, J., J.E. Craig, and S.L. Thein, Fetal hemoglobin levels in adults. Blood Rev, 1994. 8(4): p. 213-24.
- Bradai, M., et al., Hydroxyurea can eliminate transfusion requirements in children with severe beta thalassemia. Blood, 2003.
- Kohli-Kumar, M., et al., Use of hydroxyurea and recombinant erythropoietin in management of homozygous beta 0 thalassemia. J Pediatr Hematol Oncol, 2002. 24(9): p. 777-8.
- Lo, L. and S.T. Singer, Thalassemia: current approach to an old disease. Pediatr Clin North Am, 2002. 49(6): p. 1165-91, v.
- Shamsi, T.S., et al., Peripheral blood stem cell transplantation in children with beta-Thalassemia major. J Coll Physicians Surg Pak, 2003. 13(4): p. 204-6.