
Clinical Evaluation of Hemoglobinopathies: Part II. Structural Changes
In the previous issue of the Warde Report, I reviewed hemoglobin production and the problems that occur in the Thalassemias where production of specific globin chains is reduced. The present article reviews the most common and most clinically significant structural alterations that occur and what type of information we need in the clinical laboratory to optimize their detection.
Background on Normal Hemoglobin Structure
Hemoglobin molecules are composed of two pairs of globin chains, each containing a heme group at its core. During fetal life, the most common molecule is hemoglobin F (HbF) that contains two alpha chains and two gamma chains ![]() (Table 1). In adults, the predominant hemoglobin molecule, hemoglobin A (HbA) contains two alpha chains and two beta chains
(Table 1). In adults, the predominant hemoglobin molecule, hemoglobin A (HbA) contains two alpha chains and two beta chains ![]() . The normal adult also has between 2.3 and 3.5% of hemoglobin A2 (HbA2)
. The normal adult also has between 2.3 and 3.5% of hemoglobin A2 (HbA2) ![]() , and may have up to 2% HbF in circulating erythrocytes.
, and may have up to 2% HbF in circulating erythrocytes.
Table 1. Normal Ranges of Adult Hemoglobins
| Hemoglobin | Globin Chain Composition | Adult Concentration* |
| HbA | 96-98% | |
| HbA2 | 2.3-3.5% | |
| HbF | <2% |
| * | Adult concentrations are usually attained by 6-8 months of age. |
General Overview of Structural Hemoglobinopathies
Most structural abnormalities are caused by a single mutation in the genetic code resulting in the substitution of one amino acid. Occasionally, the genetic background for the structural hemoglobinopathy is more complex. There are a few abnormal hemoglobins that result from two amino acid substitutions and some others that are the product of a mutation that removes a stop codon producing an exceptionally long and usually unstable hemoglobin molecule.
As of October, 2003, 875 structural hemoglobin variants of hemoglobin have been reported in the literature (see Globin Gene Server web site listed in references for details). Fortunately, most variants do not produce disease. Patients who carry these “silent” variants have no hematological or clinical abnormalities due to the presence of the variant. They are usually detected when a hemoglobin electrophoresis is performed during the evaluation of an anemia that resulted from some other cause. Some clinically silent hemoglobin variants can produce changes in the CBC. For instance, hemoglobins that have high affinity for oxygen will result in an increased erythrocyte count and elevated hemoglobin concentration, although the patients with these findings are clinically well. In contrast, individuals with low oxygen affinity hemoglobins, such as HbF will have a lower erythrocyte count and a lower level of hemoglobin. However, just as with high oxygen affinity hemoglobins, patients who are homozygous for hereditary persistence of fetal hemoglobin are clinically well.
Clinically Significant Hemoglobin Variants
Unfortunately, some hemoglobin variants can have significant effects both clinically and hematologically. The most common clinically significant structural variants and their original geographical associations are listed in Table 2. However, in countries such as the United States with a diverse immigrant population, these and many other variants may be seen. This is why I encourage you to provide a clinical history of the country of origin of affected individuals and their family members on the requisition. This information can be of help in narrowing down the type of structural abnormality when some of the more unusual variants are detected by the sensitive high-pressure liquid chromatography system that is used to evaluate all samples submitted to Warde Medical Laboratory.
Table 2. Common Clinically Significant Structural Variants
| Hemoglobin | Original Geographic Association | Amino Acid Substitution | % Present in Heterozygotes |
| S | Africa | 40% | |
| C | Africa | 40% | |
| E | Asia | 30% | |
| G Philadelphia* | Africa | 20, 30, 40% ** | |
| D Los Angeles*** | Europe/India | 40% | |
| O Arab | Africa | 40% | |
| Lepore# | Mediterranian | 10-15% |
| * | Other G variants exist which may be |
| ** | HbG Philadelphia is an |
| *** | Also known as D Punjab (same mutation) |
| # | Not a mutation, Lepore is the product of the indicated crossover during meiosis. |
Hemoglobin S
![]() is the most common clinically significant hemoglobin abnormality that we see in the United States. Sickle trait is present in 8% and homozygous sickle disease occurs in 1 of every 600 African Americans (Steinberg, 2001). The first description of a patient with sickle cell anemia was published in 1910 when Herrick reported the sickle-shaped erythrocytes in the peripheral smear of a young man. As shown in Table 2, HbS has the normal glutamic acid in the sixth position of the beta chain replaced by valine due to a point mutation in the DNA. Deoxygenation of HbS causes it to polymerize resulting in the sickling characteristic of this disease. The sickled erythrocytes are more rigid and are more likely to stick to the endothelial surfaces which causes them to obstruct small blood vessels. This same mutation (valine in the sixth position of the beta chain) is also present in a rare hemoglobin variant, HbC (Harlem), that also sickles under conditions of deoxygenation.
is the most common clinically significant hemoglobin abnormality that we see in the United States. Sickle trait is present in 8% and homozygous sickle disease occurs in 1 of every 600 African Americans (Steinberg, 2001). The first description of a patient with sickle cell anemia was published in 1910 when Herrick reported the sickle-shaped erythrocytes in the peripheral smear of a young man. As shown in Table 2, HbS has the normal glutamic acid in the sixth position of the beta chain replaced by valine due to a point mutation in the DNA. Deoxygenation of HbS causes it to polymerize resulting in the sickling characteristic of this disease. The sickled erythrocytes are more rigid and are more likely to stick to the endothelial surfaces which causes them to obstruct small blood vessels. This same mutation (valine in the sixth position of the beta chain) is also present in a rare hemoglobin variant, HbC (Harlem), that also sickles under conditions of deoxygenation.
Sickle Cell Anemia (HbSS) is a severe disease that presents clinically shortly after the HbF levels fall to adult levels (at about 6 months of age) (Keren 1st article in this series). During the first few months of life, the child is spared from symptoms of this disease because HbF, high oxygen affinity hemoglobin, interferes with sickling. Indeed, some therapies currently in use try to increase the HbF concentration in children and adults with HbSS to decrease their need for transfusion. It is important to diagnose sickle cell anemia during the first few months of life to help the parents and clinician understand symptoms of vaso-occlusive attacks that begin when the HbF level falls. With HbSS, after the first year of life, patients usually have 90-95% HbS in their erythrocytes (the remainder being HbA2 and HbF). At this concentration of HbS, sickling occurs under conditions of deoxygenation such as with stasis, infection or acidosis.
Because of the sickling, the patients suffer a chronic hemolytic anemia and the usual 120 day half life of erythrocytes falls to a mere 20 days for patients with sickle disease. As a result, the hemoglobin concentration of sickle disease patients who are not in crisis is typically in the range of 6-10 g/dl. The chronic hemolysis results in a rapid production of erythrocytes and a CBC that contains reticulocytes, occasional nucleated erythrocytes, target cells and sickle cells. As the spleen becomes compromised with repeated infarctions, Howell-Jolly bodies are also seen. Several factors can lead to crises in sickle cell disease (Table 3). Anything that leads to hypoxia will promote sickling. Therefore, patients with respiratory compromise due to infection or chronic respiratory diseases are prone to develop sickle crisis.
In addition, individuals with sickle cell disease should be cautioned about traveling to higher altitudes where the lower atmospheric pressure provides less oxygen per breath. Sickling itself is a relatively slow process such that even though erythrocytes may begin to sickle, as they pass back to the lung, they may be reoxygenated and revert to the normal biconcave shape. However, if there is vascular stasis, the erythrocytes may be delayed enough that there is permanent membrane damage and they become irreversibly sickled. Both cold and dehydration result in vasoconstriction and hemoconcentration resulting in increased propensity to sickling. Lastly, fever and low pH promote sickling in these erythrocytes. Therefore, when individuals with sickle disease contract an infection, they must be carefully monitored for development of sickle crisis. Also, individuals with sickle cell disease who develop diabetes must carefully control their blood sugar to avoid both the complications of diabetes and the sickling effects of acidosis.
The presence of HbS elevates the measurement of HbA2 when high-pressure liquid chromatography (HPLC) is used to measure it. This results because a glycated portion of HbS elutes from the column at the same place that HbA2 does.
Table 3. Factors Leading to Sickle Cell Crisis
| Factors | |
| Hypoxia | Dehydration |
| Vascular Stasis | Fever |
| Cold | Acidosis |
Sickle Cell Trait (HbAS) is a benign condition. The CBC is normal with no sickle forms seen on a smear of peripheral blood made from a freshly drawn blood sample. When performed by gel electrophoresis, the typical case contains about 40% HbS and 60% HbA. However, when the measurement of HbS is performed by high performance liquid chromatography (HPLC), the percentage of S is slightly lower than by gel techniques because the glycated fraction of HbS migrates in the HbA region. This makes no difference for the detection and characterization of the molecule, but will help to explain the slightly lower percentage of HbS when measured by HPLC in patients with HbAS. At concentrations of oxygen present in the normal circulation, sickling does not occur in these heterozygotes. However, at high altitude, or other conditions with extremely low oxygenation such as severe pneumonia, sickling may occur. In addition, painless hematuria (usually from the left kidney) occurs in about 5% of these patients and some have difficulty concentrating urine (likely reflecting local hypoxia in the kidney).
Sickle Trait and Thalassemia. Combinations of sickle cell trait with other conditions can either increase or decrease the percentage of HbS that is present. For instance, patients who are doubly heterozygous for sickle / ![]() Thalassemia will behave clinically like patients with sickle disease. The only functioning beta chain product they have results in HbS. This possibility may be suspected when we correlate the HPLC pattern with the CBC results. Patients with HbSS with no Thalassemia have a normal MCV and severe anemia. Those with doubly heterozygous sickle /
Thalassemia will behave clinically like patients with sickle disease. The only functioning beta chain product they have results in HbS. This possibility may be suspected when we correlate the HPLC pattern with the CBC results. Patients with HbSS with no Thalassemia have a normal MCV and severe anemia. Those with doubly heterozygous sickle / ![]() Thalassemia produce no beta chain product from one chromosome and the MCV is low. In this situation, HbA2 is also higher than usual. Family studies or molecular techniques could be used to confirm this impression.
Thalassemia produce no beta chain product from one chromosome and the MCV is low. In this situation, HbA2 is also higher than usual. Family studies or molecular techniques could be used to confirm this impression.
Patients who are doubly heterozygous for sickle trait and one of the milder forms of ![]() Thalassemia will produce HbA, HbS and HbA2. The percentage of HbS is higher than the percentage of HbA in contrast to the usual findings in sickle cell trait (Figure 1). Depending on the type of
Thalassemia will produce HbA, HbS and HbA2. The percentage of HbS is higher than the percentage of HbA in contrast to the usual findings in sickle cell trait (Figure 1). Depending on the type of ![]() Thalassemia, the percentage of HbA may be as little as 5% to as high as 30%. The clinical severity of this condition is inversely related to the percentage of HbA present.
Thalassemia, the percentage of HbA may be as little as 5% to as high as 30%. The clinical severity of this condition is inversely related to the percentage of HbA present.
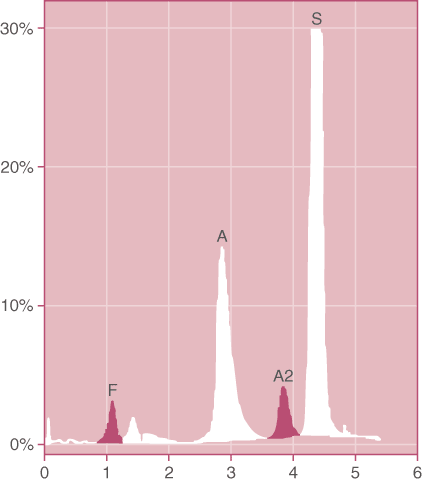
Figure 1. Doubly Heterozygous HbS / Thalassemia. The amount of HbS is much greater than the amount of HbA in this patient who had not received a transfusion. In addition, the MCV was low with a normal RDW. (HPLC pattern)
Sickle Trait and Thalassemia. Deletions of ![]() -genes are relatively common, single deletions occurring in 30% of African Americans. Patients who are doubly heterozygous for sickle /
-genes are relatively common, single deletions occurring in 30% of African Americans. Patients who are doubly heterozygous for sickle /![]() Thalassemia will produce less HbS than patients with uncomplicated sickle trait. As discussed in the first article in this series, there are 4
Thalassemia will produce less HbS than patients with uncomplicated sickle trait. As discussed in the first article in this series, there are 4 ![]() -genes on chromosome 16 and that deletion of 1, 2 or 3 of these genes results in progressive decrease in the amount of
-genes on chromosome 16 and that deletion of 1, 2 or 3 of these genes results in progressive decrease in the amount of ![]() -globin chains available to couple with the
-globin chains available to couple with the ![]() -globin chains. Because the normal
-globin chains. Because the normal ![]() -globin chains combine better with the limited number of
-globin chains combine better with the limited number of ![]() -globin chains than does the variant
-globin chains than does the variant ![]() -globin, the percentage of HbS formed is proportionately lower than usual. With two or three alpha gene deletions, the percentage of HbS in the heterozygote can fall as low as 20%. A similar effect is seen in sickle trait individuals who suffer from iron deficiency.
-globin, the percentage of HbS formed is proportionately lower than usual. With two or three alpha gene deletions, the percentage of HbS in the heterozygote can fall as low as 20%. A similar effect is seen in sickle trait individuals who suffer from iron deficiency.
Sickle Trait and Hereditary Persistence of Fetal Hemoglobin (HPFH). HbF is a high oxygen affinity hemoglobin and patients who are homozygous for HbF are clinically well. In the African American the most common form of HPFH results from deletion of both the ![]() and
and ![]() genes on the short arm of chromosome 11. Patients who are doubly heterozygous for HbS/HbF produce 60-70% HbS and 20-30% HbF, the remainder of the hemoglobin accounted for by HbA2. Because the HbF ameliorates the sickling effect and is evenly distributed in the erythrocytes in this condition, sickling does not occur.
genes on the short arm of chromosome 11. Patients who are doubly heterozygous for HbS/HbF produce 60-70% HbS and 20-30% HbF, the remainder of the hemoglobin accounted for by HbA2. Because the HbF ameliorates the sickling effect and is evenly distributed in the erythrocytes in this condition, sickling does not occur.
Hemoglobin C
![]() is relatively common in the United States with HbC trait present in 2% of African Americans. As shown in Table 2, HbC has the normal glutamic acid in the sixth position of the beta chain (the same position where a substitution occurs for HbS) replaced by lysine due to a point mutation in the DNA. HbC is less soluble than HbA and will form large oblong intracellular crystals. In splenectomized patients, the erythrocytes containing crystals are more numerous. The crystallization process is not related to the degree of oxygenation.
is relatively common in the United States with HbC trait present in 2% of African Americans. As shown in Table 2, HbC has the normal glutamic acid in the sixth position of the beta chain (the same position where a substitution occurs for HbS) replaced by lysine due to a point mutation in the DNA. HbC is less soluble than HbA and will form large oblong intracellular crystals. In splenectomized patients, the erythrocytes containing crystals are more numerous. The crystallization process is not related to the degree of oxygenation.
Homozygous Hemoglobin C (HbCC) causes a mild hemolytic anemia, possibly due to the crystal formation. Before newborn screening programs, because the patients are usually asymptomatic, the condition was often not diagnosed until adulthood. Because the spleen removes the crystal-containing erythrocytes, splenomegaly is commonly present in adults with this condition. The half-life of HbCC erythrocytes is only 32 days. The peripheral blood smear shows microcytosis, target cells and a few pyknotic spherocytes. With HbCC, after the first year of life, patients usually have 90-95% HbC in their erythrocytes (the remainder being HbA2 and HbF). The presence of HbC may elevate the measurement of HbA2 when HPLC is used to measure it, but to a much lesser extent than seen with HbS.
HbC Trait (HbAC) is a benign condition. The CBC demonstrates slightly low to low-normal MCV with an occasional target cell. The typical case contains about 40% HbC and 60% HbA. As with HbS, the co-inheritance of alpha Thalassemia deletions results in a progressive decrease of the percentage of HbC as the number of deletions increase.
Doubly Heterozygous HbSC. HbSC disease occurs in about 1 in 2,500 African Americans. The concentration of HbS and HbC are approximately 50:50 (Figure 2). HbF is <2% in most cases. The presence of both HbS and HbC results in a mild amount of sickling (much less than in homozygous sickle disease or doubly heterozygous sickle/ ![]() Thalassemia) that causes a moderately severe hemolytic anemia (hemoglobin >10). While the condition is generally benign, avascular necrosis of the femoral head occurs more commonly than in sickle cell disease. Unlike HbSS where the cells have a normal MCV, in HbSC the cells have a low to low-normal MCV, but they have an increased MCHC. In addition one may see occasional target cells and crystals, but true sickle cells are rarely seen.
Thalassemia) that causes a moderately severe hemolytic anemia (hemoglobin >10). While the condition is generally benign, avascular necrosis of the femoral head occurs more commonly than in sickle cell disease. Unlike HbSS where the cells have a normal MCV, in HbSC the cells have a low to low-normal MCV, but they have an increased MCHC. In addition one may see occasional target cells and crystals, but true sickle cells are rarely seen.
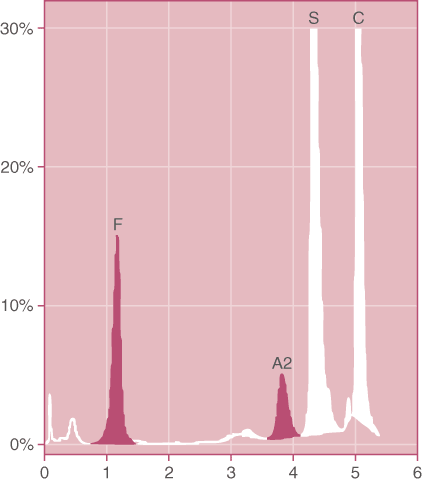
Figure 2. Doubly heterozygous Hemoglobin SC. Note the relatively large percentage of HbF that often accompanies this condition. In addition, HbA2 is elevated due to the presence of glycated HbS and HbC in this band. (HPLC pattern)
Other Double Heterozygous Hemoglobinopathies. Fishleder and Hoffman pointed out that the clinical severity of homozygous and doubly heterozygous hemoglobinopathies varies considerably. In some cases, such as HbSS, it is predictably a severe process. In other cases, such as HbSD, it may be surprising that a relatively severe process results while homozygous HbDD has no hematological consequences at all (Table 4).
Table 4. Relative Clinical Severity of Various Hemoglobinopathies
| Ranking of Clinical Severity | Hemoglobin |
| None | AA, DD, AG, CO, SG |
| Mild | EE |
| Mild to moderate | CC, OO |
| Moderate to severe | SC, SD |
| Severe | SS, SO |
Data modified from Fishleder and Hoffman (Lab Med 1987;18:513.)
Hemoglobin E
![]() has become much more common in the United States with the increasing number of individuals of Southeast Asian origin now living here. About 10% of these immigrants carry HbE trait. The frequency varies in Southeast Asia with the highest frequency found in Thai and Khmer regions, more moderate occurrence in Malaya, fewer in Vietnam and rare in China and Japan. As shown in Table 2, HbE has the normal glutamic acid in the 26th position of the beta chain replaced by lysine due to a point mutation in the DNA. In addition to creating a different hemoglobin structure, this results in the production of a new (cryptic) splicing site that produces a non-functional messenger RNA during some of the attempts to read the DNA. Therefore, less functional
has become much more common in the United States with the increasing number of individuals of Southeast Asian origin now living here. About 10% of these immigrants carry HbE trait. The frequency varies in Southeast Asia with the highest frequency found in Thai and Khmer regions, more moderate occurrence in Malaya, fewer in Vietnam and rare in China and Japan. As shown in Table 2, HbE has the normal glutamic acid in the 26th position of the beta chain replaced by lysine due to a point mutation in the DNA. In addition to creating a different hemoglobin structure, this results in the production of a new (cryptic) splicing site that produces a non-functional messenger RNA during some of the attempts to read the DNA. Therefore, less functional ![]() -globin chains are produced. This explains the significantly lower concentration of HbE in the heterozygote state (Table 2) and also the thalassemic picture that both heterozygotes and homozygotes for HbE display.
-globin chains are produced. This explains the significantly lower concentration of HbE in the heterozygote state (Table 2) and also the thalassemic picture that both heterozygotes and homozygotes for HbE display.
Homozygous Hemoglobin E (HbEE) is a clinically benign condition, despite the presence of noticeable hematological abnormalities in the CBC. The patients have a mild microcytic anemia, typically with about 12g/dl of hemoglobin and an MCV of about 65 fL. Despite the mild anemia, the reticulocyte count is not elevated. However, target cells are commonly seen in the peripheral blood smear. The HPLC pattern demonstrates about 98% HbE with the remainder being HbF.
HbE Trait (HbAE). This is also clinically benign with no anemia. As mentioned above, the cryptic splicing site created by the point mutation that alters the beta chain in patients with HbE results in a decreased production of these beta chains such that HbE in the heterozygote typically comprises 30% of the total hemoglobin, with the remainder being HbA. As with the HbS and HbC, the co-inheritance of alpha Thalassemia results in a progressive decrease in the percentage of HbE with 1, 2 or 3 deletions of alpha genes.
Doubly Heterozygous HbE / Thalassemia. In contrast to the benign nature of homozygous HbE (HbEE), patients who inherit both HbE and ![]() Thalassemia suffer a moderate to severe microcytic anemia with an average of only 6 g/dl of hemoglobin. Their HPLC pattern typically demonstrates about 35-70% HbE with the remainder being HbF. As discussed in the first article in this series, it is the excess free alpha chains due to the combination of HbE and
Thalassemia suffer a moderate to severe microcytic anemia with an average of only 6 g/dl of hemoglobin. Their HPLC pattern typically demonstrates about 35-70% HbE with the remainder being HbF. As discussed in the first article in this series, it is the excess free alpha chains due to the combination of HbE and ![]() Thalassemia that further damage the membranes of the erythrocytes. Patients with this condition have a clinical picture with the severity of either Thalassemia intermedia or Thalassemia major depending on whether
Thalassemia that further damage the membranes of the erythrocytes. Patients with this condition have a clinical picture with the severity of either Thalassemia intermedia or Thalassemia major depending on whether ![]() Thalassemia or
Thalassemia or ![]() Thalassemia is inherited. The most severely affected are transfusion dependent and will require chelation therapy to avoid excess iron deposition.
Thalassemia is inherited. The most severely affected are transfusion dependent and will require chelation therapy to avoid excess iron deposition.
Hemoglobin G Philadelphia
![]() , unlike the structural variants discussed thus far, is a structural variant that involves the alpha chain rather than the beta chain. HbG Philadelphia has the normal asparagine in the 68th position of the alpha chain replaced by lysine due to a point mutation in the DNA (Table 2). It has a frequency of about 1 in 5,000 African Americans, but has been reported in other ethnic groups in the Mediterranean region as well. Recall that there are four alpha genes, two on each chromosome 16. One might expect with one alpha gene mutated, that 25% of the hemoglobin would be HbG and the remainder HbA with small amounts of HbA2. However, the HbG variant is usually linked to a one alpha chain deletion thereby yielding closer to 30% HbG in many individuals. The presence of another deletion of an alpha gene on the chromosome uninvolved with the structural variant could result in an HbG as high as 40%.
, unlike the structural variants discussed thus far, is a structural variant that involves the alpha chain rather than the beta chain. HbG Philadelphia has the normal asparagine in the 68th position of the alpha chain replaced by lysine due to a point mutation in the DNA (Table 2). It has a frequency of about 1 in 5,000 African Americans, but has been reported in other ethnic groups in the Mediterranean region as well. Recall that there are four alpha genes, two on each chromosome 16. One might expect with one alpha gene mutated, that 25% of the hemoglobin would be HbG and the remainder HbA with small amounts of HbA2. However, the HbG variant is usually linked to a one alpha chain deletion thereby yielding closer to 30% HbG in many individuals. The presence of another deletion of an alpha gene on the chromosome uninvolved with the structural variant could result in an HbG as high as 40%.
Fortunately, HbG itself has no clinical consequences. The only finding of clinical interest is the mild microcytosis that is not due to the HbG, but to the associated alpha gene deletions. A couple of the laboratory findings may cause confusion. On gel electrophoresis, it migrates in the same position as HbS and could be mistaken if the laboratory does not perform another test to confirm the identity of HbS. In our laboratory we use HPLC and HbG does not share migration with HbS by this technique. Laboratories using only gel techniques can make the distinction by performing an acid gel procedure and/or an in vitro test for sickling. Another interesting feature that may be confusing on the final interpretation relates to this being an alpha chain variant. HbA2 is a combination of ![]() (Table 1). Since there are both normal and variant alpha chains in these patients, in addition to producing normal HbA2, the variant alpha chains will combine with some of the normal chains to produce a molecule termed
(Table 1). Since there are both normal and variant alpha chains in these patients, in addition to producing normal HbA2, the variant alpha chains will combine with some of the normal chains to produce a molecule termed ![]() (Figure 3). While this molecule has no clinical consequences, the presence of this molecule (about 1%) on a carefully examined gel or HPLC pattern, helps to distinguish HbG from HbD that shares its electrophoretic migration on HPLC and gel electrophoresis and which does have consequences when it occurs with sickle trait (Table 4).
(Figure 3). While this molecule has no clinical consequences, the presence of this molecule (about 1%) on a carefully examined gel or HPLC pattern, helps to distinguish HbG from HbD that shares its electrophoretic migration on HPLC and gel electrophoresis and which does have consequences when it occurs with sickle trait (Table 4).
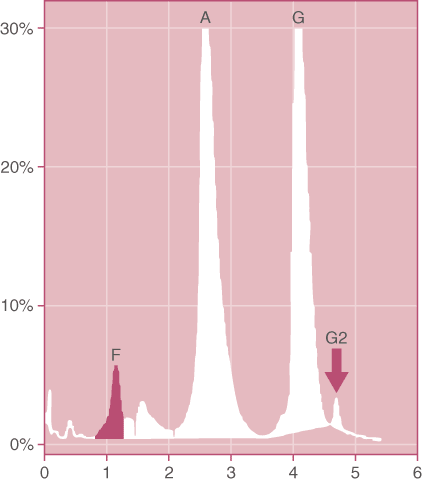
Figure 3. G-Philadelphia Trait. Note that the G2 peak is present due to the combination of the variant G-beta chain with the normal delta chains. (HPLC pattern)
Hemoglobin D Los Angeles
![]() is relatively uncommon in the United States with a frequency of about 1 in 30,000 African Americans. The same structural hemoglobin is also referred to as HbD Punjab because it has a prevalence of about 3% in individuals from India and Pakistan. In addition, it has a prevalence of about 1 in 10,000 Northern European individuals. HbD Los Angeles has the normal glutamic acid in the 121st position of the beta chain replaced by glutamine due to a point mutation in the DNA (Table 2).
is relatively uncommon in the United States with a frequency of about 1 in 30,000 African Americans. The same structural hemoglobin is also referred to as HbD Punjab because it has a prevalence of about 3% in individuals from India and Pakistan. In addition, it has a prevalence of about 1 in 10,000 Northern European individuals. HbD Los Angeles has the normal glutamic acid in the 121st position of the beta chain replaced by glutamine due to a point mutation in the DNA (Table 2).
When inherited as an isolated structural variant, or even as the rare homozygote form, HbD has no clinical or hematological consequences. However, in doubly heterozygous HbD/HbS, the HbD is able to enhance the sickling due to copolymerization with the HbS. This results in a severe sickling disorder (Table 4). Because of this, distinction between HbD and HbG is of more than academic interest. Since HbD is a beta variant, it does not produce a variant form of HbA2. Further, the percentage of HbD is considerably higher than that of HbG, except when two alpha gene deletions are present in the latter. However, in that case, the variant form of HbA2 together with microcytosis from the two alpha gene deletions would point to the correct interpretation.
Hemoglobin O Arab
![]() has the normal glutamic acid in the 121st position of the beta chain replaced by glutamine due to a point mutation in the DNA (Table 2). HbO Arab is uncommon in the United States with a frequency of about 1 in 30,000 African Americans. It is most commonly found in the Balkan Region and the Middle East. Hoyer and Kroft point out that despite its name, HbO Arab is rare among the Arab population.
has the normal glutamic acid in the 121st position of the beta chain replaced by glutamine due to a point mutation in the DNA (Table 2). HbO Arab is uncommon in the United States with a frequency of about 1 in 30,000 African Americans. It is most commonly found in the Balkan Region and the Middle East. Hoyer and Kroft point out that despite its name, HbO Arab is rare among the Arab population.
As a heterozyote, HbO Arab has no clinical or hematological consequences. Homozygote HbO Arab is associated with a mild to moderate anemia. Doubly heterozygous HbSO is associated with a sickling anemia similar in severity to sickle cell disease (Table 4). The total hemoglobin in these patients is 7-8 gm/dl. They display a prominent reticulocytosis. Clinically they resemble sickle cell disease and require transfusion. Doubly heterozygous HbO/ ![]() Thalassemia also is associated with a relatively severe microcytic anemia with a hemoglobin as low as 6 g/dl.
Thalassemia also is associated with a relatively severe microcytic anemia with a hemoglobin as low as 6 g/dl.
Hemoglobin Lepore
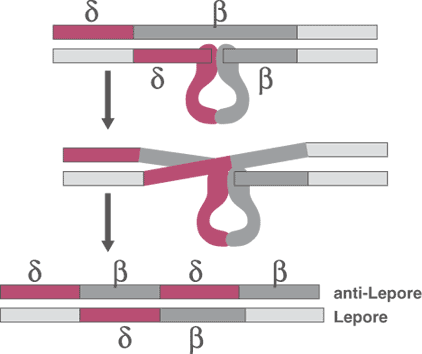
Hb Lepore is an unusual molecule that is the product of the fusion of residues 1-87 of the delta chain and residues 116-146 of the beta chain (Figure 4). This fusion results from a non-homologous crossover of the DNA from these two genes during meiosis. Hb Lepore is synthesized at a decreased rate and consequently, it gives a thalassemic picture. The heterozygote usually has 10-15% Hb Lepore (Table 2), but is a stable molecule. It occurs most frequently in patients originating from the Mediterranean region. Because of the low rate of production, heterozygotes have a low MCV and elevated erythrocyte count. Homozygotes have an anemia similar to, or occasionally more severe than Thalssemia Intermedia.
Figure 4.The gene for hemoglobin Lepore is created by a non-homologous crossover of the DNA during meiosis.
Final Comments
Hemoglobinopathies are complex because of the diversity of the mutations that can affect both the structure of the molecules and the amount of the molecules produced. This together with the environmental factors such as iron deficiency, vitamin B12 and folate deficiency and other clinical conditions that alter the CBC can make interpretation of these variants and their clinical consequences extremely difficult. In the first article of this series, I recommended providing as much clinical information to the laboratory as possible to allow us to provide the best possible interpretation. When any questions arise about the interpretation in a given case, please feel free to call our laboratory and ask to speak with the pathologist about the case.
References
- Akar, N and Ekim, M. Further notes on hemoglobin C and hematuria. Pediatr Nephrol 2002;17:72.
- Fishleder, AJ and Hoffman, GC. A practical approach to the detection of hemoglobinopathies: Part III. Lab Med 1987;18:513.
- Globin Gene Server, HbVar: a database of human hemoglobin variants and Thalassemias. http://globin.cse.psu.edu/cgi-bin/hbvar/counter (accessed May 2003).
- Hoffman, GC. The Sickling Disorders. Lab Med 1990;21:797.
- Hoyer, JD, Kroft, SH, (Ed). Color Atlas of Hemoglobin Disorders. CAP Press, 2003.
- Hoyer, JD. Laboratory approach to the diagnosis of hemoglobin S and HbS-related disorders. ASCP Teleconference. Spring 2000.
- Keren, DF. Clinical evaluation of hemoglobinopathies. Part I. Thalassemia. The Warde Report 2003 ;14, no. 2:1.
- Koduri, PR, et at. Hemoglobin S-C disease revisited: Clinical study of 106 adults. Am J Hematol 2001;68:298.
- Mayo, MM and Samuel, SM. Diagnosing hemoglobin SC. Lab Med 1999;30:98.
- Powars, DR, et al. Outcome in hemoglobin SC disease: a four-decade observational study of clinical, hematologic, and genetic factors. Am J Hematol 2002;70:206.
- Romana, M. et al. Hemoglobin Sickle-Lepore: an unusual case of sickle cell disease. Acta Haematol 1997;98:170.
- Schmugge, M, et al. Stroke in hemoglobin (SD) sickle cell disease with moyamoya: successful hydroxyurea treatment after cerebrovascular bypass surgery. Am J Hematol 2001;97:2165.
- Steinberg, MH. Modulation of fetal hemoglobin in sickle cell anemia. Hemoglobin. 2001;28:195.
- Wajeman, H, et al. Abnormal hemoglobins: laboratory methods. Hemoglobin 2001;25:169.
- Wang, JCY, et al. Hemoglobinopathies: three illustrative case studies. Clin Invest Med 1994;17:461.
- Weatherall, DJ. Hemoglobin E beta-thalassemia: an increasingly common disease with some diagnostic pitfalls. J Pediatr 1998;132:765.