
FLAER-Based Flow Cytometry Analysis of Peripheral Blood for Paroxysmal Nocturnal Hemoglobinuria (PNH)
Background

Paroxysmal nocturnal hemoglobinuria (PNH) is an unusual disorder that, in its classic form, is marked by the triad of intravascular hemolysis (and resulting hemoglobinuria), peripheral blood cytopenias due to decreased marrow function, and intravascular thrombosis, often involving unusual anatomic sites. The full-blown syndrome of classic PNH is rare, but more subtle variants of the disease are relatively common in the setting of other disorders of the bone marrow, particularly aplastic anemia (AA) and certain low-risk types of myelodysplastic syndromes (MDS).[1] As such, laboratory assessment for PNH has become a common tool for the workup of patients with Coombs-negative (i.e. non-antibody-mediated) hemolysis, acquired AA, low-risk MDS, unexplained peripheral blood cytopenias, or clinically atypical thrombosis.[1]
Initial descriptions in the 19th century observed “intermittent hematinuria” in affected patients, later found to correlate with the in-vitro phenomenon of enhanced hemolysis in a weakly acidic environment due to a non-antibody mediated process.[2] Further work revealed that blood cells in patients with PNH are deficient in proteins that normally inhibit formation of the complement membrane attack complex (MAC), resulting in increased susceptibility of red blood cells to complement-mediated lysis.[3]
The more basic pathophysiology of PNH involves the absence of glycophosphoinositol (GPI) on the cell membrane. GPI acts as an “anchor” molecule for the attachment of certain proteins (collectively termed GPI-linked proteins) to the cell surface. As a result, affected cells are deficient in all GPI-linked proteins, although only certain ones (such as CD55 and CD59 in the case of red blood cells) are directly connected to observed clinical signs and symptoms.[4]
Although the disease itself is named for the effect of GPI deficiency on red blood cells, PNH originates at the level of the bone marrow hematopoietic stem cell, and therefore blood cells of all lineages are affected.[3] The fundamental molecular lesion of PNH is an acquired somatic mutation of the PIG-A gene, which codes for the GPI anchor molecule. Cells that acquire this mutation also appear to acquire a growth advantage over normal cells, resulting in the emergence of a so-called “PNH clone,” or a subset of marrow cells – each derived from the same mutated hematopoietic stem cell – that lack expression of GPI-linked proteins. (The basic reasons for this growth advantage are not entirely clear, and many mechanisms have been proposed, including differential response of PNH cells to positive and negative regulators of cell growth, relative resistance of PNH cells to immune surveillance within the bone marrow, and concomitant activation of genes involved in cell growth.[2,4]) PNH is often referred to as a “clonal but nonmalignant” condition of the bone marrow. The size of the PNH clone relative to non-affected cells in the blood and bone marrow correlates with the presence or absence of classic PNH signs and symptoms.[2]
| Clinically, PNH occurs in three main forms: [1] |
| Classic PNH, with clinically manifest hemolysis or thrombosis. As mentioned above, classic PNH is rare, but is associated with the presence of a sizable PNH-type clone within the blood and bone marrow, often comprising 10% or more of blood cells. A review by Parker purports that the threshold between subclinical and clinical PNH is reached when detectable PNH granulocyte clones reach levels of approximately 20 to 25%. [3] |
| PNH-type clones present within the context of another defined bone marrow disorder such as acquired aplastic anemia (AA) or certain low-risk types of myelodysplastic syndromes (MDS). As noted above, the lack of GPI-linked proteins may provide a selective advantage to PNH-type cells in marrow failure syndromes such as AA or MDS. It is reported that approximately 50 to 60% of AA patients and approximately 15 to 20% of low-risk MDS patients harbor some level of PNH-type clones using high sensitivity methods.[3] Furthermore, the presence of PNH-type clones in both AA and MDS has been associated in some studies with increased likelihood of response to immunosuppressive therapy. |
| Subclinical PNH clones that occupy a small percentage of cells in blood and marrow but that are not associated with concomitant marrow pathology. |
There has been renewed focus on PNH diagnosis in recent years, largely due to the development of more sensitive methods for detection, favorable responses to immunosuppressive therapy in AA and MDS patients who harbor PNH clones, and the emergence of an FDA approved therapy for classic PNH in the form of eculizumab, a monoclonal antibody directed against the C5 component of the complement cascade, and an effective inhibitor of the complement-induced hemolysis and thrombosis characteristic of this disorder. [5]
Historically, PNH was diagnosed with labor-intensive and insensitive tests designed to demonstrate enhanced susceptibility to complement-mediated red blood cell (RBC) lysis.[2,6] The sucrose hemolysis (or “sugar water”) test exploited the tendency of RBCs to absorb complement in a low ionic strength environment, with subsequent measurable complement-induced in-vitro hemolysis in samples from affected individuals. Positive sucrose hemolysis tests were often followed by an acidified serum (or “Ham’s”) test for confirmation, again based on the principle that complement binds to RBCs at weakly acidic pH. A fairly sizable PNH clone was needed in order to react as positive, and positive tests were potentially seen in conditions other than PNH. For these reasons, these tests have largely given way to flow cytometry-based analyses as a more sensitive and specific approach for detection of PNH clones.
Flow Cytometric Diagnosis of PNH Using Fluorescent-Labeled Aerolysin (FLAER)
Flow cytometry is capable of detecting the presence of a PNH-type clone within a peripheral blood sample to a remarkable threshold of sensitivity, often detecting clones comprising less than .01% (fewer than 1 in 10,000) of total cells analyzed.[6] After the initial description of the feasibility of using flow cytometry of peripheral blood for assessment of GPI-linked proteins in the workup of PNH,[7] early clinical applications focused on assessment of CD55 (decay accelerating factor — DAF) and CD59 (membrane inhibitor of reactive lysis – MIRL),[8] since both regulators of assembly of the complement membrane attack complex (MAC) on the surface of RBCs, and both are involved directly with the hemolysis characteristic of PNH. However, subsequent studies demonstrated relatively low sensitivity for the detection of PNH clones (in both erythrocytes and leukocytes) using these two markers, and a lack of consistent separation between populations expressing and lacking these proteins.[6]
Many GPI-linked proteins have been targeted in flow cytometric PNH screening, but some markers are less useful due to variable expression patterns across the spectrum of normal blood cells, or varied levels of expression in different stages of ontogeny or activation states of certain blood cell types.[6] For example, decreased expression of CD16 may be seen in immature granulocytes, which may be present in circulation due to infection or inflammatory conditions, and may lead to a false-positive PNH screen if CD16 is used as a primary marker. Likewise, absence of CD14 may be meaningful evidence of loss of GPI-linked proteins on mature monocytes but not on monocyte precursors or on granulocytes, raising the possibility of false positive PNH screens due to suboptimal gating strategies in flow cytometric analysis.
In 2000, Brodsky and colleagues advanced the field of PNH diagnosis by exploiting the ability of bacterial derived aerolysin and proaerolysin to bind selectively and with high affinity to the GPI-anchor itself.[9] They demonstrated the effectiveness of using a fluorescently-labeled aerolysin reagent (FLAER) to detect PNH clones. Since then, consensus guidelines have been developed by the International Clinical Cytometry Society (ICCS) that recommend the use of at least two GPI-targeted reagents – one preferably being FLAER – in flow cytometry-based PNH screening.[6]
For initial detection of PNH-type populations, screening of granulocyte and monocyte populations is overall more sensitive than RBC screening, since active hemolysis or transfusion may markedly reduce the number of circulating RBCs that lack GPI-linked proteins, potentially resulting in false-negative PNH screens or in the underestimating of PNH clone size.[4,6] At Warde Medical Laboratory, our FLAER-based PNH screen (test code PNHF) employs a 5-color, single-tube flow cytometry approach that uses antibodies to three markers (CD15, CD45, CD64) for selection (gating) of granulocyte and monocyte populations, and two reagents (FLAER and anti-CD157) for detecting GPI-deficient (PNH-type) populations. This approach is in line with ICCS consensus guidelines,[6] and the use of CD157 in particular is a novel approach published by Sutherland and colleagues that streamlines WBC PNH screening.[10] CD157 is a GPI-linked protein that is consistently expressed at relatively high levels by both granulocytes and monocytes, eliminating the need for additional lineage-specific GPI-linked protein targets, and avoiding the lower sensitivity of markers that show lower or more variable levels of expression in normal populations.[10]
This approach to PNH screening allows for the rapid recognition of normal (Type I) cell populations (Figure 1), partially GPI-deficient (Type II) cell populations (Figure 2), or fully GPI-deficient (Type III) cell populations (Figure 3) to a threshold of less than 0.01% of total cells analyzed.
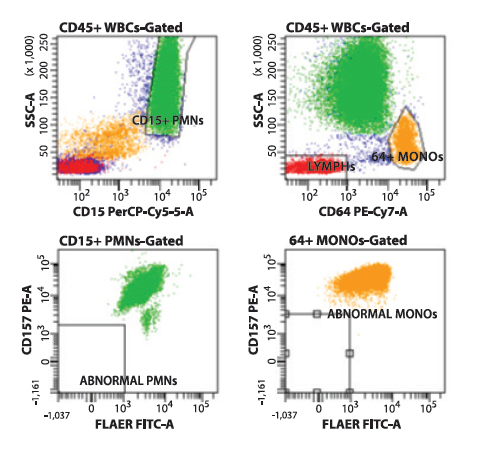
Figure 1. Flow cytometry histograms showing normal (Type I) cells on CD157/FLAER PNH analysis. Neutrophils (green) are selected based on CD15 expression and side angle light scatter. Monocytes (yellow) are selected based on CD64 expression and side angle light scatter. The selected populations are analyzed using FLAER and anti-CD157 reagents, demonstrating no cellular events in the FLAER/CD157-deficient quadrant (lower left) of each gated histogram.
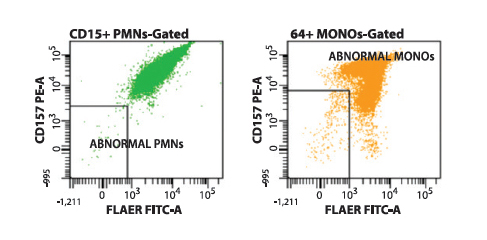
Figure 2. Flow cytometry histograms showing the presence of a type II (partially GPI-deficient) PNH-type clone on CD157/FLAER PNH analysis. Neutrophils (green) and monocytes (yellow) each show varied levels of deficiency in CD157 expression and binding of FLAER.
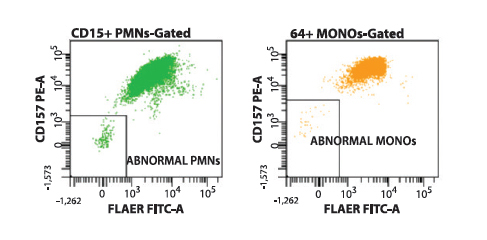
Figure 3. Flow cytometry histograms showing the presence of a type III (fully GPI-deficient) PNH-type clone on CD157/FLAER PNH analysis. Neutrophils (green) and monocytes (yellow) each show distinct, clustered populations deficient in expression of CD157 and binding of FLAER.
Summary
The detection of PNH-type cell populations has taken on renewed significance with the development of highly sensitive flow cytometry-based assays, and with the advent of effective therapies for PNH and related disorders. In addition to its usefulness in the workup of Coombs-negative intravascular hemolysis or hemoglobinuria, screening for the presence of PNH-type clonal populations in peripheral blood has become an important part of the clinical evaluation in the setting of unexplained cytopenias, acquired AA, low-risk MDS, and atypical patterns of thrombosis. For patients undergoing initial evaluation of these conditions, the Warde FLAER-based peripheral blood PNH screen (test code: PNHF) offers a rapid, efficient, and highly sensitive option.
References
- Parker C, Omine M, Richards S, Nishimura J, et al: Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood 2005; 106(12):3699-3709.
- Young NS, Maciejewski JP: Genetic and environmental effects in paroxysmal nocturnal hemoglobinuria. J Clin Invest 2000; 106(5):637-641.
- Parker CJ: Paroxysmal nocturnal hemoglobinuria. Curr Opin Hematol 2012; 19:141-148.
- Parker CJ: The pathophysiology of paroxysmal nocturnal hemoglobinuria. Exp Hematol 2007; 35:523-533.
- Kelly RJ, Hill A, Arnold LM, Brooksbank GL, et al: Long-term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival. Blood 2011; 117(25):6786-6792.
- Borowitz MJ, Craig FE, DiGiuseppe JA, Illingworth AJ, et al: Guidelines for the diagnosis and monitoring of paroxysmal nocturnal hemoglobinuria and related disorders by flow cytometry. Cytometry Part B2010; 78B:211-230.
- Van der Schoot CE, Huizinga TWJ, Van’t Veer-Korthof ET, Wijmans R, et al: Deficiency of glycosyl-phosphatidylinositol-linked membrane glycoproteins of leukocytes in paroxysmal nocturnal hemoglobinuria, description of a new diagnostic cytofluorometric assay. Blood 1990; 76(9):1853-1859.
- Hall SE, Rosse WF: The use of monoclonal antibodies and flow cytometry in the diagnosis of paroxysmal nocturnal hemoglobinuria. Blood 1996; 87(12):5332-5340.
- Brodsky RA, Mukhina GL, Li S, Nelson KL, et al: Improved detection and characterization of paroxysmal nocturnal hemoglobinuria using fluorescent aerolysin. Am J Clin Pathol 2000; 114:459-466.
- Sutherland DR, Acton E, Keeney M, Davis BH, Illingworth A: Use of CD157 in FLAER-based assays for high-sensitivity PNH granulocyte and PNH monocyte detection. Cytometry Part B 2014; 86B:44-55.