
Testing for Increased Susceptibility to Clotting. What? When? Why?
Two million people in the USA are treated annually for deep venous thrombosis (DVT), and 200,000 die. Many more are placed on medications such as oral contraceptives, hormone replacement and tamoxifen which are known to predispose to thrombosis. A number of tests identify additional congenital and acquired factors predisposing to thrombosis. However, most of these “thrombophilic” (clot-predisposing) conditions are quite rare. As a result, the diagnostic yield of these tests in general is low, the potential for false positive values is high and the cost to the health care system for performance of these tests is relatively large. This publication attempts to outline a conservative, cost and yield conscious approach to this type of testing and the limitations of these tests.
Pre (Analytic) Testing Issues:
Virchow, the father of modern pathology, first observed that diseases predisposing to thrombi could be categorized into blood vessel disorders, heart (circulatory) disorders and blood disorders. The perceived need for extensive testing for the identification of thrombophilic blood conditions is frequently obviated by the fact that an underlying etiology is evident to the treating physician in a large proportion of cases (see Table 1).
Table 1. Common Conditions Associated with Thrombosis
| Physical Conditions | Disease Related | Iatrogenic |
| Pregnancy | Cancer | Surgery |
| Obesity | Cardiac valvular disease | Medications |
| Trauma | Heart failure | |
| Inactivity | Collagen vascular disease | |
| Systemic disease |
Despite evidence suggesting that additional occult laboratory testing identified conditions in these patients that may also be present, there is little perceived need for the laboratory to identify additional factors.
Laboratory testing of patients at the time of presentation with thrombus has limitations. DVT, pulmonary and arterial emboli are serious medical conditions, and lead to an “acute phase” type inflammatory response, which impairs laboratory test interpretation by temporarily altering baseline values (either up or down, depending on the analyte). Coagulation factors, such as VIII, can double in an acute phase phase response. This abnormality can result in activated Protein C resistance in the absence of Factor V Leiden, as a result of excessive factor VIII binding to the added Protein C reagent (see next section). Recent thrombosis, pregnancy and estrogen therapy reduce Protein S levels up to 50%, overlapping with values seen with heterozygous deficiency.
Treatment of thrombosis consists of heparin (unfractionated or low molecular weight), and coumadin. Heparin therapy reduces antithrombin III 30-40%, nearly to levels seen in antithrombin III deficiency states, and coumadin reduces vitamin K dependent Factors, those being Factors 2, 7, 9, 10, and natural anti-coagulants Protein C and Protein S. Reduction in vitamin K dependent factor levels with coumadin therapy can result in a falsely negative lupus anticoagulant test with some reagents. Chronic antibiotic therapy alters bowel flora resulting in vitamin K deficiency. Heparin half-life is prolonged in renal failure (Table 2).
Table 2: Common Interferences in Testing Due to
Acute Thrombosis or Thrombosis Therapy
| AGENT | TEST | RESULT ON TEST |
| “Acute phase” response | FVIII activity APCR Protein S (Activity and Ag) | Elevated from baseline Falsely positive Decreased from baseline up to 40% |
| Heparin | Antithrombin III activity | Decreased 30-40% |
| Coumadin | Factors 2,7,9,10, Protein C,S | Reduced due to Vitamin K inhibition |
| Wide spectrum antibiotics | Protein C,S, Factors 2,7,9,10 | Reduced due to Vitamin K deficiency |
| Thrombin Inhibitors (Refluden®, esp.) | Virtually all | Unpredictable |
For the above reasons, it is more advantageous to delay thrombophilic testing until 2-3 months after an acute event, one week after discontinuing heparin, and two weeks after discontinuing coumadin.
Factor V Leiden (FVL) and Activated Protein C Resistance (APCR):
Factor V Leiden is a genetic variant resulting from one amino acid substitution (Arginine to Glutamine) at sequence #506 of the Factor V gene. It was first identified in the Netherlands because laboratory testing for anticoagulation there is standardized and much of the research work is performed at a single central Leiden laboratory. It is relatively common (3-8%) among Western Europeans. It is believed that the presence of the gene confers a survival advantage by reducing hemorrhage at childbirth.
Protein C is a vitamin K dependent natural anticoagulant which acts by binding to and inactivating factors V and VIII. It is presumed that patients with heterozygous or homozygous factor V Leiden develop thrombi because their factor V is not functionally deactivated as quickly by Protein C, resulting in excessive and prolonged thrombosis (Figure 1).

Figure 1: Factor V Leiden does not interact with
Protein C and is therefore not deactivated.
The APCR test, first described by Dahlbeck, now consists of a PTT performed using a mixture of patient plasma and factor V deficient plasma (which enhances sensitivity by reducing the factor V concentration), with and without added “activated” (thrombin treated) Protein C (Table 3).
Table 3: Activated Protein C Resistance Test
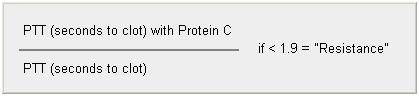
Those patients whose PTT does not prolong appropriately after adding the anticoagulant Protein C are termed “resistant” to Protein C and are likely to carry the mutation.
Physiological changes during pregnancy increase Protein C resistance. Acute phase reactions lead to elevated factor VIII, which can bind the additional Protein C reagent and result in a false positive activated Protein C resistance assay. Patients with a prolonged PTT due to antiphospholipid antibody (lupus anticoagulant) can produce borderline APCR results because their PTT is prolonged and does not change significantly with added Protein C. For these above reasons, definitive diagnosis of FVL is made by DNA testing, performed on specimens identified by the more inexpensive APCR assay.
Factor V Leiden heterozygote carriers are six- to eightfold more likely to have venous thrombosis than the general population. Further, homozygous patients are predisposed to develop thrombosis from 30-50 x the general population. Some literature indicates that these patients have an increased risk of (the more serious) arterial thrombosis while the majority of the literature does not support this conclusion. There is no consensus advocating treatment of patients with this trait in the absence of a history of thrombi, or treating them longer or to a higher anticoagulation level. Despite the synergy of Factor V Leiden with thrombophilic conditions like estrogen therapy, oral contraceptives and smoking, there is no perceived necessity to identify Factor V Leiden carriers prior to embarking on these therapies or in smokers. In short, we look for Factor V Leiden because it is commonly present in patients with thrombi and because, if identified in a patient with a thrombus, it argues for lifetime anticoagulation if no other factors are evident to explain the clot.
Antiphospholipid Antibodies (aka lupus anticoagulants):
Coagulation reactions occur in the test tube and in patients on phospholipid rich surfaces (called thromboplastins in the laboratory, and platelets in patients). Antibodies to phospholipid molecules commonly occur in patients receiving certain medications (e.g. hydalazine and procainamide) and those with infections. They also occur in patients with autoimmune diseases (from where the adjective “lupus” comes). In most patients with lupus anticoagulants, however, no predisposing condition, medication, infection, or autoimmune disorder is clinically evident. Lupus anticoagulants are identified by neutralizing a prolonged phospholipid dependent coagulation test with an extra dose of phospholipids or platelets (which consistent mainly of phospholipids). The laboratory simply does a before and after phospholipid addition clotting time (PTT or DRVTT), and looks at the ratio (Table 4).
Table 4: Criteria for positive Lupus Anticoagulant
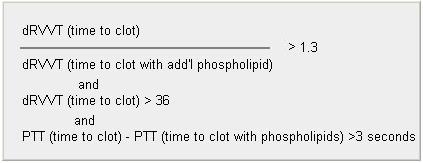
If the ratio with phospholipid addition is increased, and the initial value is prolonged, the laboratory calls the test positive. Many laboratory’s, including Warde, screen patients using the DRVVT, but demand a positive PTT confirmation in addition. Laboratory’s became aware of antiphospholipid antibodies by doing clotting times utilizing clotting agents which contained phospholipids (like PTT) and by performing serologic assays like VDRL and RPR, which may detect these antibodies. While lupus anticoagulants prolong PTT’s, protimes (PT) are usually unaffected, because there is usually too much phospholipid in the PT reagent for the antibody to interfere.
Another more direct way to identify antiphospholipid antibodies is to perform an enzyme-linked immunosorbent assay (ELISA) If antiphospholipid antibodies are present, a color change in the reaction measures the amount of the antibody. As mentioned above, syphilis RPR and VDRL tests contain phospholipids antigens and may give positive reactions on sera that contain antiphospholipid antibodies. Because the phospholipid source for these tests were historically beef heart (cardiolipin), anti-phospholipid antibodies are sometimes called “anti-cardiolipin antibody” (ACL). However, not all patients with lupus anticoagulants have anticardiolipin antibodies. Since some patients with anticardiolipin antibodies show no lupus anticoagulant activity, it is recommended that when testing for an antiphospholipid antibody, clinicians should request both lupus anticoagulants and anticardiolipin antibodies.
The term “lupus anticoagulant” is paradoxical. While it does describe what happens in the test tube (prolong PTT), it is the opposite of what happens in the patient (thrombosis). In vivo, we believe these antibodies bind to naturally occurring anticoagulant proteins, inactivate them, and promote thrombi. Some of the thrombi are arterial, but most are venous. Large and small vessels can be affected, especially the latter on the lower extremities, resulting in a peculiar net-like rash known as livido reticularis. Involvement of the placenta in obstetric patients results in miscarriage, placental infarction and fetal heart block. It is not known if the ability to inhibit in vitro clotting, binding of cardiolipin, or immunoglobulin type is more important to causing thrombi in vivo, so laboratories test for all of these characteristics. The present hypothesis is that the ability of an antibody to bind a certain protein, or small group of proteins, could explain the clinical phenomena. In particular, several phospholipids like Beta2 glycoprotein I, prothrombin, Protein S, Protein C, and annexin V (in obstetric patients) have all been implicated as the target of an antiphospholipid antibody that results in thrombosis. Unfortunately, for every positive piece of literature, there are several refuting it. Unique testing issues, as well as lack of laboratory standardization, may be the reason. However, at this time, the consensus of the literature is to screen for antiphospholipid antibodies with both lupus anticoagulant (clot based) plasma assays and IgG, IgA and IgM anticardiolipin assays, until additional clinical research sorts out the pathogenesis of thrombosis in these patients.
Prothrombin G20210A (not 90210, like the TV show):
The same Leiden group also identified a variation in 2-3% of Western Europeans in the prothrombin (Factor II) molecule resulting from a single nucleotide substitution (guanine to adenine at sequence #20210). The resulting elongated thrombin molecule has about 30% more activity than normal in cleaving fibrinogen, which results in an increased risk for venous, but not arterial, thrombosis. Because the normal range for thrombin activity overlaps to a great degree with that of individuals having the G20210A trait, laboratories identify the G20210A trait with DNA testing, not a clot based assay. The significance of the mutation is debated. It is a weaker risk factor for thrombosis (about twice that of the general population) than Factor V Leiden mutation. There is no indication to treat an asymptomatic carrier and no increased risk of the more dangerous arterial thrombi, except perhaps in neonates. Homozygotes are quite rare but may have up to 6 times the risk of thrombosis. Despite this, because the abnormal DNA is relatively easy to identify and because the trait is relatively common in patients with thrombi (5-10%), routine screening panels for thrombophilia contain this test.
Antithrombin III (ATIII):
Heterozygous (50% of normal) deficiency of Antithrombin III, which is quite rare, results in a moderately severe risk state for venous and arterial thrombosis. Typically, these patients present before age 50, so the appropriate strategy is to screen for this in the same patients in whom you screen for C and S deficiency, and those with arterial thrombi. Unfractionated (standard) heparin reduces ATIII levels in normal patients by 1/3 at least, causing overlap with the deficiency state. Wait a week after heparin therapy ends before testing for ATIII.
Homocysteine:
Few laboratory tests better exemplify the paradigm of laboratory testing (first underused, then overused, then underused again) than homocysteine. Elevated levels as a risk factor for thrombosis, especially arterial, were first hypothesized as a result of autopsy findings in children with elevated homocysteine levels. Retrospective clinical studies document an increased risk for venous and arterial thrombosis with elevated plasma values of homocysteine (>12).
However, the following issues cloud its use. Some studies indicate that measurement should be done after methionine loading, while most indicate simple fasting. Despite the use of folate, Vitamins B12, and/or B6 to reduce plasma levels, the majority of studies indicate that reducing the plasma homocysteine level does not reduce the thrombosis risk, suggesting that elevation is an unrelated phenomenon. Because of the latter observation, few centers presently advocate routine testing for homocysteine in thrombophilic screening. Further, in the patient in whom homocysteine screening is considered, genotypic studies are not warranted, and screening should consist of fasting homocysteine measurement only.
Protein C and S Deficiency:
These defects are rare. As noted above, both of these vitamin K dependent natural anticoagulant proteins bind (and inactivate) Factors V and VIII, thus inhibiting thrombosis. Homozygotes (0% activity) for these factors die in utero. Heterozygotes (with approximately 50% of normal activity) have a highly variable condition. Some individuals have no or rare thrombi while others may have arterial and venous thrombi commencing in childhood. Neonatal death due to widespread thrombi in Protein C deficiency is well documented. The literature suggests looking for these patients only if thrombi occur before age 45. These proteins are sensitive to coumadin, estrogen, and pregnancy, all of which result in reduced values in non-deficient people. Lupus anticoagulants reduce Protein S activity but not antigenic mass (concentration).
Patients with Protein C and S deficiency usually do not make enough Protein C or Protein S and therefore both activity and antigenic mass (concentration) are proportionately decreased. Some patients however, have decreased activity because of functionally defective molecules, and therefore have a normal antigenic mass.
For Protein C, the initial screen is an activity (clot) based assay. However, the same is not true of Protein S. Elevated Factor VIII levels, lupus anticoagulants, thrombin inhibitors and acute phase reactions can all interfere with total Protein S clotting activity, the “gold standard” assay. A better screening assay for Protein S deficiency is free Protein S antigen because it is more precise than total Protein S clotting activity. Because Protein S activity assays require functional capability of the molecule which can be lost in transit to the laboratory and because the assay is sensitive to factors other than Protein S, we recommend Free Protein S antigen as the initial screening assay for Protein S deficiency. We recommend that all low Protein S values be confirmed by functional activity when the patient is not actively ill, on estrogens or coumadin, or pregnant. If a patient has a family history of Protein S deficiency, and the free Protein S antigen is normal, a total Protein S activity assay is warranted to detect the rare family having normal amounts of dysfunctional Protein S (so called type II deficiency).
Other Thrombophilic Conditions:
A number of other plasma protein deficiency states and platelet membrane Protein deficiency states have been identified in select families which are predisposed to thrombosis. They include plasminogen activator inhibitor deficiency, Heparin cofactor II deficiency, glycoprotein IIb/IIIa (CD41/61) anomalies, and Factor VII and Factor XIII genetic polymorphisms and platelet function abnormality (so called “sticky platelets”). Furthermore, elevated activity of several coagulation factors (especially Factor VIII), above normal is associated with an increased likelihood of future thrombosis. Despite these associations, the consensus of the literature is that routine screening for these conditions, in the absence of a specific family history of these unusual conditions, is not indicated.
Conclusions:
| A | We recommend the following “panel” of tests for patients with thrombosis, to be drawn several weeks after the acute event, if possible:Activated Protein C resistance (Factor V Leiden DNA to be reflexively added if positive).Lupus anticoagulant screen, with confirmation by the addition of phospholipids if the screening assay is prolonged.Anticardiolipin antibodies (IgG, A and M)Prothrombin G20210A DNA |
| B | In patients with no predisposing conditions, and a family history of thrombosis or with presentation before age 50 or with arterial thrombi, an additional panel consisting of fasting homocysteine, Protein C activity, free Protein S antigen, and ATIII activity, can be considered. Further testing beyond this should be categorized as research. |
| C | Abnormalities identified during acute illness, or while the patient is on heparin or coumadin, must be confirmed at another time when the patient is in their usual state, has discontinued heparin for one week or coumadin for two weeks, and is not pregnant or taking estrogens. |
| D | Assay of coagulation factors (especially Factor VIII) for elevated levels as a thrombophilic condition is not indicated because of the lack of interlaboratory standardization of these assays. |
| E | The identification of antibodies against selected phospholipids (e.g. Beta2 glycoprotein I, Annexin V, prothrombin, Protein S, and others) is an area of research without proven clinical utility at present (but keep in touch). |
| F | Retesting for disappearance of previously identified anticardiolipin antibody or lupus anticoagulants 3-6 months after detection should be considered before a thrombophilic condition is diagnosed. |
Reference
Archives of Pathology and Laboratory Medicine; Vol. 126, No. 11, November 2002, College of American Pathologists Consensus Conference XXXVI: Diagnostic issues in Thrombophilia.