
Thrombophilia (Hypercoagulable States)
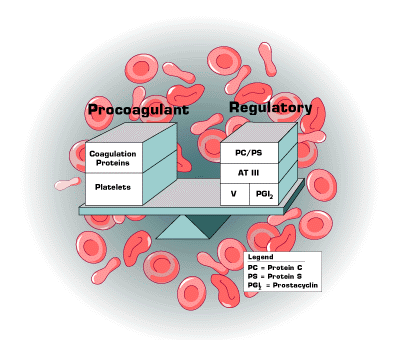 |
In 1856, Rudolf Virchow, a German pathologist, proposed a hypothesis to explain the pathogenesis of thrombosis. He suggested there were three primary causes of venous and arterial thrombosis: stasis, injury to the vessel wall and abnormalities in the circulating blood. Subsequently, numerous investigators contributed the concept of a hemostatic balance between fibrin formation and fibrin dissolution (Figure 1).
Although many families with a predisposition to thrombosis were subsequently described, it wasn’t until 1965 that a Norwegian physician, O. Egberg reported a family with a partial antithrombin deficiency (originally termed antithrombin III) (1). He carefully analyzed the family and noted the association of the antithrombin deficiency with venous thromboembolic events. Members of the family who did not have antithrombin deficiency were asymptomatic. In this seminal paper, he also proposed the term, thrombophilia. Thrombophilia may be defined as hereditary or acquired conditions which predispose individuals to thromboembolic events. Following Egberg’s publication, additional hereditary causes of thrombophilia were identified in the 1980s including protein C and protein S deficiencies (Table 1) (2,3). Protein C, protein S, and antithrombin are proteins involved in the downregulation of coagulation. Deficiencies of these proteins result in an increased generation of thrombin and a predisposition to thrombosis.
| History of Thrombophilia | ||
| 1856 | Virchow | Thrombosis Hypothesis |
| 1965 | Egberg | Antithrombin Deficiency |
| 1981 | Griffith | Protein C Deficiency |
| 1984 | Comp | Protein S Deficiency |
| 1993 | Dahlback | APC-R |
| 1994 | Bertina | Factor V Leiden |
| 1996 | Poort | Prothrombin 20210A |
Bjorn Dahlback’s description of Activated Protein C-Resistance (APC-R) in 1993 dramatically changed the diagnosis and management of venous thrombotic events (4). Dahlback described a large family from Southern Sweden who demonstrated thrombosis in males and females throughout several generations (autosomal dominant pattern of inheritance). When he evaluated the family utilizing the available assays to identify patients with hereditary thrombophilia (e.g., protein C, protein S, antithrombin assays), all laboratory results were within appropriate normal reference intervals. In addition, he performed a simple experiment adding Activated Protein C (APC) to patient and normal plasmas. He observed a significant prolongation of the APTT in the normal plasma following the addition of APC (APC inactivates factor Va and factor VIIIa decreasing available thrombin) (1 ). (Figure 2) In contrast, plasmas from the affected family showed a lack of significant prolongation of the APTTs. Dahlback concluded there was an abnormality in the protein C/protein S regulatory system. Subsequently, this abnormality was identified as a single amino acid substitution in one of the substrate proteins for APC: Factor V. This mutation was later characterized by Bertina and colleagues at the University of Leiden (factor V Q506, factor V Leiden) (5). The rapidity with which the genetic mutation was identified following the phenotypic observation of APC-R is illustrative of the dramatic shift to gene-based diagnosis of diseases. Dahlback subsequently reported that as many of 15% of the population in Southern Sweden carried the factor V Leiden (FVL) gene. The identification of FVL significantly changed the way clinicians and laboratories approach the diagnosis of thrombophilia.
Evaluation of Venous Thrombosis in 1998
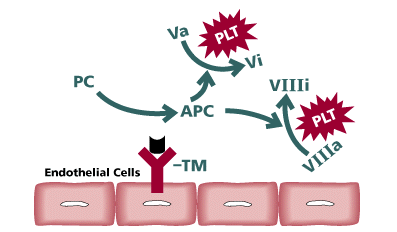 | ||
| Legend | ||
| TM = Thrombomodulin PLT = Activated Platelets PC = Protein C APC = Activated Protein CThe protein C/protein S pathway is critical in the regulation of hemostasis. When thrombin is generated in vivo, it is inhibited by antithrombin. Also,Thrombin may bind to an endothelial cell receptor thrombomodulin. thrombomodulin will inhibit the procoaguland properties of thrombin. As the name implies, it”modulates” thrombin activities. The complex of TM-T is capable of converting protein C to active protein C (APC). APC in the presence of protein S will inactivate factor Va and factor VIIIa localized on phospholipid membranes. |
With the discovery of FVL, clinicians were able to increase the positive results of thrombophilia screens by 20-25% in patients presenting with their first event. Prior to 1993, patients with a family history of venous thrombosis who were screened with the available tests (protein C, protein S, antithrombin) were found to be positive for a genetic disorder in 8-10% of the cases. With the availability of APC-R/FVL testing, patients presenting with a first thrombotic event are now found to have a demonstrable abnormality in approximately 25-30% of the cases. The frequency of the FVL in the North American Caucasian population is estimated at 4-5%. Consequently, FVL/APC-R testing should be a part of evaluating any patient of European descent who presents with a venous thrombotic event. The risk of recurrent thromboembolism is significantly higher in carriers of FVL than in patients without this hereditary abnormality. When evaluating selected patients with a history of two or more thromboembolic events, approximately 40-45% of these individuals will be found to have FVL. FVL patients with a previous veinous thrombotic event have a high frequency of recurrence if they are not adequately anticoagulated.
A polymorphism in the prothrombin gene (20210 G ![]() A) was described in 1996 (6 ). This mutation occurs in the 3 untranslated portion of the prothrombin gene and results in increased levels of circulating prothrombin (>115% IU/dl). This mutation is found primarily in people of European descent. Approximately 2-3% of this population is found to have the 20210 G
A) was described in 1996 (6 ). This mutation occurs in the 3 untranslated portion of the prothrombin gene and results in increased levels of circulating prothrombin (>115% IU/dl). This mutation is found primarily in people of European descent. Approximately 2-3% of this population is found to have the 20210 G ![]() A polymorphism. In patients presenting with deep venous thrombosis (DVT)s, approximately 6% are positive for the 20210 G
A polymorphism. In patients presenting with deep venous thrombosis (DVT)s, approximately 6% are positive for the 20210 G ![]() A polymorphism.
A polymorphism.
In addition to hereditary disorders predisposing to venous thrombosis, there are many acquired conditions which amplify hereditary thrombophilia or represent significant risk factors in the absence of genetic risk (Table 2).
| Venous Thrombosis: Risk Factors† | ||
| Genetic (Hereditary)* | Acquired/Environmental | Mixed |
| Factor V Leiden (FVR506Q) | Age | |
| MTHFR** (Polymorphisms) | History of Prior Thrombosis | |
| Factor II 20210 A | Surgery/Trauma | |
| Protein S Deficiency | Extended Bed Rest | |
| Protein C Deficiency | Malignancy | |
| Antithrombin Deficiency | Oral Contraception | |
| Dysfibrinogenemias | Hormonal Replacement Therapy Antiphospholipid Antibodies Pregnancy/Puerperium Myeloproliferative Disorders Obesity/Inactivity | |
| †Modified from Rosendaal Seminars in Hematology 34:171-187, 1997 *Listed in order of frequency; note, there is a relatively low incidence of venous thrombosis in Asian and African populations **MTHFR = Methylenetetrahydrofolate Reductase |
Acquired and Combined Risk Factors
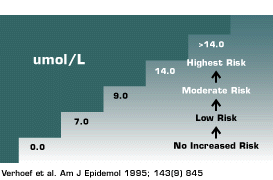 | ||
| Legend | ||
| Most laboratories report “normal ranges” of homosyteine from 7-14 umol/L. However, there is an increased risk of vascular complications when patients have homocystine levfels within the “normal range”. |
Kilmer McCully, a pathologist, first suggested there was a connection between increased homocysteine levels and arterial disease approximately 30 years ago. Recently hyperhomocystinemia has emerged as a risk factor for both venous and arterial thrombosis (7,8). Although the association of increased homocysteine levels with vascular thromboembolic disease remains a topic of some controversy, increasingly clinicians are measuring homocysteine levels. With the introduction of new technologies to measure plasma homocysteine (e.g., ELISA, liquid chromatography, and synthetic substrates), homocysteine assays in the near future will cost less and will become widely available. There appears to be a gradient relationship between plasma homocysteine levels and vascular disease (Figure 3).
In many cases, elevated homocysteine is due to underlying dietary deficiencies particularly of vitamins B6, B12, and folate. This year, the FDA approved folate fortification of cereals and breads. This public health initiative may address a significant percentage of patients who have mild hyperhomocystinemia. As a result, the incidence of neural tube defects and vascular disease would logically decrease.
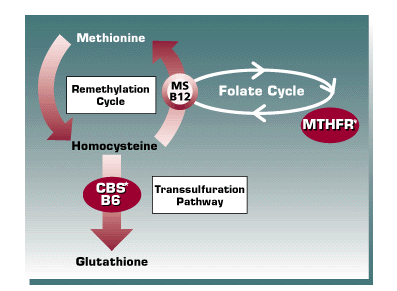 | ||
| Legend | ||
| Enzymes: 1. MTHFR = methylenetetrahydrofolate reductase 2. MS = methionine synthase 3. CBS = cystathionine ß-synthaseVitamines: 1. Folate 2. Vitamin B12 3. Vitamin B6There are three key enzymes involved in homocysteine metabolism: MTHFR, MS,CBS, and three vitamins, which are cofactors for the enzymatic reactions. |
Two frequent polymorphisms have been identified in the gene for methylenetetrahydrofolate reductase (MTHFR) (Figure 4). The first defect described identified a polymorphism (C677T; labile MTHFR) which occurs in approximately 40-45% of the Caucasian population. The frequency of homozygosity for this mutation approaches 10%. In the heterozygous state, homocysteine levels are usually within the normal reference interval. However, homozygous patients may have increased homocysteine levels. When combined with deficiencies of B vitamins, heterozygous C677T patients often have mildly increased homocysteine levels. A second polymorphism of the MTHFR gene has recently been described: 1298 (A ![]() C) (9). This mutation often occurs in the “trans” position together with labile MTHFR C677T polymorphism. The 1298 (A
C) (9). This mutation often occurs in the “trans” position together with labile MTHFR C677T polymorphism. The 1298 (A ![]() C) mutation occurs with a frequency of 33% in the Dutch population. One would expect a similar percentage in other Europeans. Combined heterozygosity for MTHFR 677 (C
C) mutation occurs with a frequency of 33% in the Dutch population. One would expect a similar percentage in other Europeans. Combined heterozygosity for MTHFR 677 (C ![]() T) and MTHFR 1298 (A
T) and MTHFR 1298 (A ![]() C) results in hyperhomocystinemia and decreased plasma folate levels.
C) results in hyperhomocystinemia and decreased plasma folate levels.
Levels of factor VIII activity exceeding 150 IU/dL have been linked to venous thrombosis (10). In some cases, this appears to be hereditary. When evaluating factor VIII levels as a risk factor for DVT, it is important to correct for an “acute phase reaction” Utilizing a factor VIIIC to fibrinogen ratio will help resolve this issue.
Antiphospholipid Antibodies (APA)
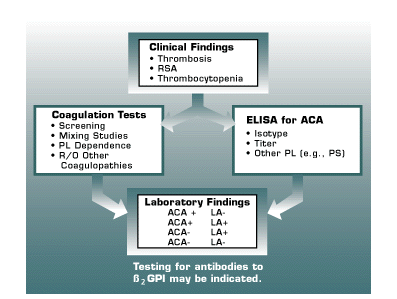
Lupus anticoagulants (LA) and anticardiolipin antibodies (ACA) are the most common causes of acquired thrombophilia. Approximately 8-14% of venous thromboembolic events are secondary to antiphospholipid antibodies (APA). Thirty percent of systemic lupus erythematosus patients are found to have underlying ACA and/or LA. However, the vast majority of patients with LA/ACA do not have SLE. The laboratory evaluation of APA requires a coordinated effort to perform both LA and ACA testing (Figure 5). The criteria required to establish the diagnosis of LA have been published (SSC Subcommittee Lupus Anticoagulants/Phospholipid-dependent Antibodies) (11). Patients with LA are at greater risk for thrombosis than patients who are only ACA positive. Several studies suggest a level of greater than 40 GPL units for ACA represents an increased risk factor for venous thrombosis. However, it is important to test these patients on more than one occasion to verify persistence of ACA (8-10 weeks after initial positive results).
 | ||
| Legend | ||
| RSA = Recurrent Spontaneous Abortions PL = Phospholipids PS = Phosphatidylserine ß2GPI = Beta 2 Glycoprotein I (a physiologic anticoagulant)Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus anticoagulants: and update. thromb haemost 74:1185-1190, 1995. |
Recently, another ELISA assay has been introduced to detect antibodies to ß2 Glycoprotein I. ß2 Glycoprotein I is a physiologic anticoagulant which is the true antigenic target for antibodies measured in ACA assays. Although ACA testing is sensitive, it is not particularly specific (many transiently positive ACA results are due to infections). Antibodies to ß2 Glycoprotein I have greater specificity in identifying patients at risk for thrombosis. Consequently, patients with moderate to high titer ACA (e.g., >40 GPL units) should be evaluated with anti-ß2 Glycoprotein I testing. ACA and anti-ß2 Glycoprotein I testing are ELISA-based assays.
Recent studies have emphasized patients who present with thrombosis secondary to APA are at very high risk for developing recurrent events if they are not adequately anticoagulated. In many cases, this requires life-long anticoagulation (INR 2.5-3.0) (12).
Evaluation of Arterial Thrombosis
Historically, patients with accelerated atherosclerosis or arterial thromboembolic events have been evaluated primarily utilizing lipid profiles. However, with recent advances, one can make a strong argument to evaluate for other risk factors predisposing to thrombosis (Table 3). Lipoprotein(a) has been identified as a risk factor for arterial thromboembolic events. Other risk factors include elevated levels of factor VIII (see above) and genetic polymorphisms of factor VII. Evaluation of plasma homocysteine and related gene polymorphisms are also found in patients with accelerated arterial disease. Recently, a polymorphism of Glycoprotein IIb/IIIa platelet receptor has been identified as a risk factor for accelerated coronary artery disease (13). In addition, APA testing is indicated in arterial thromboembolic disease. Often APA positive patients present with transient ischemic attacks or stroke. Many of these patients have been misdiagnosed as multiple sclerosis. Antibodies to oxidized LDL have been associated with arterial thromboembolic disease.
| Arterial Thrombosis: Risk Factors | ||
| Genetic (Hereditary)* | Acquired/Environmental | Mixaed |
| Hypercholesterolemia | Anticardiolipin Antibodies | |
| Anti Oxidized LDL | ||
| Hypertriglycerides | Diet ( | |
| Diabetes Mellitus | Smoking | |
| Hypertension | ||
| Hemochromatosis (2 Genes) | Infections (Chlamydia, CMV, etc.) | |
| PLA2 (Glycoprotein IIa/IIIb) | Social Class | |
| Factor VII Polymorphisms | Body Mass Index | |
| Hyperhomocystinemia (Variant MTHFRs) | ||
| PAI-1 Polymorphisms | ||
| Gender | ||
| Family History of Accelerated ASHD | ||
| Legend:HDL=High density lipoproteins CMV=Cytomegaloinclusion virus |
Summary
The laboratory diagnosis of thrombophilia has changed dramatically within the last five years. In the case of recurrent venous thrombosis, approximately 60% of patients are now identified as having hereditary and/or acquired risk factors. This information is extremely helpful in long-term patient management and, in the case of genetic thrombophilia, the extended family. This is particularly important in factor V Leiden where oral contraception considerably increases the relative risk of thrombosis (heterozygous FVL plus oral contraception 30-fold increase, homozygous FVL plus oral contraception 90 to 100-fold increase) (14). Correctly identifying hereditary risk factors together with appropriate family evaluation and counseling is indicated. The informed patient and physician working together will be able to effectively manage thrombophilia and prevent subsequent thrombotic events.
References
- Egeberg O. Inherited antithrombin deficiency causing thrombophilia. Thromb Haemost (STUTTG) 13:516-530, 1965.
- Griffin JH, Evatt B, Zimmerman TS et al. Deficiency of protein C in congenital thrombotic disease. J Clin Invest 68:1370-1373, 1981.
- Comp PC, Nixon RR, Cooper DW et al. Familial protein S deficiency is associated with recurrent thrombosis. J Clin Invest 74:2082-2088, 1984.
- Dahlback B, Carlsson M, Svensson PJ. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: prediction of a cofactor to activated protein C. Proc Natl Acad Sci USA 90:1004-1008, 1993.
- Bertina RM, Koeleman BPC, Koster T et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 369:64-66, 1994.
- Poort SR, Rosendaal FR, Reitsma PH et al. A common genetic variation in the 3í untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood 88:3698-3703, 1996.
- McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol 56:111-128, 1969.
- Welch GN, Loscalzo J. Homocysteine and atherothrombosis. N Engl J Med 338:1042-1050, 1998.
- Van der Put NMJ, Gabreels F, Stevens EMB et al. A second common mutation in the methylenetetrahydrofolate reductase gene: an additional risk factor for neural tube defects? Am J Hum Gen 62:1044-1051, 1998.
- Koster T, Blann AD, Briet E et al. Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep vein thrombosis. Lancet 345:152-155, 1995.
- Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus anticoagulants: an update. Thromb Haemost 74:1185-1190, 1995.
- Triplett DA. Antiphospholipid protein antibodies: laboratory detection and clinical relevance. Thromb Res 78:1-31, 1995.
- Weiss EJ, Bray PF, Tayback M et al. A polymorphism of a platelet glycoprotein as an inherited risk factor of coronary thrombosis. N Engl J Med 334:1090-1094, 1996.
- Vandenbrouke JP, Koster T, Briet E. et al. Increased risk of venous thrombosis in oral contraceptive users who are carriers of factor V Leiden mutation. Lancet 344:1453-1457, 1994.